
Cancer research has entered an era in which molecular profiling is central to biomarker discovery, therapeutic target identification, and precision oncology. While genomic and transcriptomic studies have revealed many cancer-associated alterations, DNA and RNA data alone cannot fully explain how tumors grow, metastasize, evade immune surveillance, or respond to treatment. Oncoproteomics addresses this gap by profiling cancer-associated proteins at scale—including their abundance, activity states, post-translational modifications, interactions, and spatial distribution—to connect molecular alterations with cancer phenotypes. This article introduces the scientific value of oncoproteomics, summarizes major mass spectrometry-based technologies, and discusses how proteomics supports cancer biomarker discovery, therapeutic target prioritization, and drug-response research.
1. WHAT IS ONCOPROTEOMICS AND WHY DOES IT MATTER?
Oncoproteomics is a branch of cancer proteomics that systematically studies cancer-associated proteins and protein-level regulatory events. Unlike genomics, which identifies DNA-level alterations, or transcriptomics, which measures RNA expression, oncoproteomics focuses on the functional molecular layer that more directly reflects cellular activity. This includes changes in protein abundance, activity states, post-translational modifications, interactions, localization, and pathway regulation (Dutta et al., 2023).
This protein-centered perspective is important because cancer phenotypes are often shaped by mechanisms that cannot be fully inferred from genes or transcripts alone. A tumor may carry a genomic alteration without corresponding pathway activation, while another tumor may display strong signaling activity through post-transcriptional or post-translational regulation. Similarly, mRNA abundance does not always correlate with protein abundance or protein activity. By measuring proteins more directly, oncoproteomics provides a closer view of the molecular processes that drive tumor growth, metastasis, immune evasion, and therapy resistance (Aebersold and Mann, 2016; Mani et al., 2022). These insights can support the early discovery of biomarker candidates and therapeutic hypotheses within a discovery-to-validation workflow in cancer research.
Therefore, oncoproteomics serves as a complementary layer within cancer multi-omics research. Genomics helps define potential molecular causes, transcriptomics captures gene-expression programs, and proteomics shows how these alterations are expressed at the protein and pathway levels. Together, these layers provide a more complete understanding of tumor biology and support more rigorous research into cancer biomarkers, therapeutic targets, and drug-response mechanisms (Hristova and Chan, 2019; Haga et al., 2023).
2. CORE TECHNOLOGIES USED IN ONCOPROTEOMICS
Modern oncoproteomics is driven primarily by mass spectrometry-based workflows, especially liquid chromatography-tandem mass spectrometry (LC-MS/MS). Depending on the research question, these workflows can be designed for global protein quantification, post-translational modification analysis such as phosphoproteomics and glycoproteomics, proteogenomics, or spatially informed protein profiling.
2.1 Quantitative LC-MS/MS Proteomics
Quantitative LC-MS/MS proteomics is the foundation of most oncoproteomics studies because it enables large-scale measurement of protein abundance across tumor tissues, cell models, body fluids, and other cancer-related samples. In a typical bottom-up proteomics workflow, proteins are enzymatically digested into peptides, separated by liquid chromatography, and analyzed by tandem mass spectrometry. Depending on the research goal, label-free quantification, isobaric labeling, data-dependent acquisition, or data-independent acquisition strategies can be used to compare protein expression patterns across biological groups. In cancer research, this approach helps identify dysregulated pathways, molecular subtypes, tumor-associated protein signatures, and candidate biomarkers. Its main strength is broad proteome coverage, but careful sample preparation, cohort design, and downstream validation are essential for reliable biological interpretation (Aebersold and Mann, 2016).
 workflow for proteomics_1780562768_WNo_765d629.webp)
Figure 1. Liquid chromatography tandem mass spectrometry (LC-MS/MS) workflow for proteomics. Image reproduced from Haga et al., 2023, Cancer Science, 114(5), 1783–1791.
2.2 Post-Translational Modification Proteomics
Post-translational modification proteomics adds a functional layer to oncoproteomics by examining how proteins are chemically modified after translation. These modifications can alter protein activity, stability, localization, interactions, and signaling output, often without changing total protein abundance. Phosphoproteomics is particularly important in cancer research because phosphorylation reflects kinase signaling and pathway activation, which are closely linked to tumor growth, metastasis, and drug response. Glycoproteomics, as another major PTM-focused approach, investigates cancer-associated glycosylation changes that may influence cell adhesion, immune recognition, invasion, and biomarker specificity (Pinho and Reis, 2015). By mapping modification sites, phosphorylated peptides, glycopeptides, or site-specific proteoforms, PTM proteomics can reveal tumor-relevant protein states that conventional abundance-based proteomics may miss. However, enrichment strategy, site localization confidence, quantitative reproducibility, and independent validation are critical for robust interpretation (Mani et al., 2022).
2.3 Single-Cell and Spatial Proteomics
Single-cell and spatial proteomics extend oncoproteomics beyond conventional bulk measurements. Bulk proteomics measures an averaged protein signal from the analyzed sample, which can obscure heterogeneity when tumor tissue contains mixed cancer cells, stromal cells, immune cells, vascular components, necrotic regions, or spatially distinct tumor areas. Single-cell proteomics aims to resolve protein-level variation among individual cells, helping researchers investigate rare cell states, therapy-resistant subpopulations, and cell-to-cell differences within tumors. Spatial proteomics preserves tissue context by linking protein expression or protein-related molecular features to defined anatomical regions. This is especially valuable for studying tumor-stroma interactions, immune infiltration, invasive fronts, and regional pathway activity. Although sensitivity, throughput, and standardization remain technical challenges, these approaches provide important tools for understanding cancer as a heterogeneous and spatially organized disease (Gatto et al., 2023).
Different Oncoproteomics Approaches and Their Applications
| Approach | What It Measures | Research Value | Key Limitation |
|---|---|---|---|
| Quantitative proteomics | Protein abundance across tumor and control samples | Biomarker discovery, molecular subtyping, pathway analysis | Requires independent validation and careful control of sample heterogeneity |
| Phosphoproteomics | Phosphorylation sites and kinase signaling | Pathway activation, drug-response mechanisms, target prioritization | Enrichment efficiency and site localization confidence affect interpretation |
| Glycoproteomics | Cancer-associated glycoforms and glycoproteins | More specific biomarker hypotheses and altered cell-surface biology | Clinical translation requires robust targeted assays |
| Proteogenomics | Integration of DNA, RNA, protein, and PTM layers | Connects genomic alterations with functional protein consequences | Requires matched multi-omics data and rigorous computational analysis |
| Spatial or single-cell proteomics | Cell-state or tissue-context protein patterns | Tumor heterogeneity and microenvironment studies | Lower throughput and higher technical complexity |
3. CANCER BIOMARKER DISCOVERY WITH ONCOPROTEOMICS
Oncoproteomics supports cancer biomarker discovery by identifying protein-level features that differ between tumor and non-tumor states, molecular subtypes, metastatic patterns, or treatment-response groups. Compared with genomics or transcriptomics alone, proteomic profiling can capture functional changes that are closer to tumor behavior, such as altered pathway activity, immune-related protein signatures, metabolic rewiring, and modified protein forms. In practical biomarker research, this means that tumor tissues, matched adjacent tissues, plasma, serum, urine, extracellular vesicles, or other clinically relevant samples can be analyzed to discover candidate markers for diagnosis, prognosis, recurrence risk assessment, therapy response prediction, or molecular stratification.
A strong example is the colorectal cancer study by Wang et al., which used an integrated extracellular vesicle (EV) proteomics workflow to discover and validate non-invasive diagnostic biomarkers. The researchers profiled EV proteins from both colorectal tumor tissues and plasma samples using DIA-MS, combining tissue-derived disease signals with circulating EV protein information. In the discovery phase, tissue and plasma EV proteomes were integrated to identify 21 candidate biomarkers with consistent disease-associated changes.
The study then moved beyond discovery proteomics by using PRM-MS to verify candidate proteins in plasma EV samples and ELISA to validate selected proteins in larger cohorts. This staged workflow ultimately narrowed the candidates to three EV proteins—HNRNPK, CTTN, and PSMC6—which were used to build the ColonTrack diagnostic model. Across 1,272 participants, ColonTrack distinguished colorectal cancer from non-cancer cases and showed strong performance for early-stage colorectal cancer detection, with the study reporting a combined AUC above 0.97, sensitivity of approximately 0.94, and specificity of approximately 0.93. This example illustrates how oncoproteomics can move from broad protein discovery to targeted verification and validation-oriented biomarker development, while still requiring further clinical evaluation before routine diagnostic use (Wang et al., 2025).
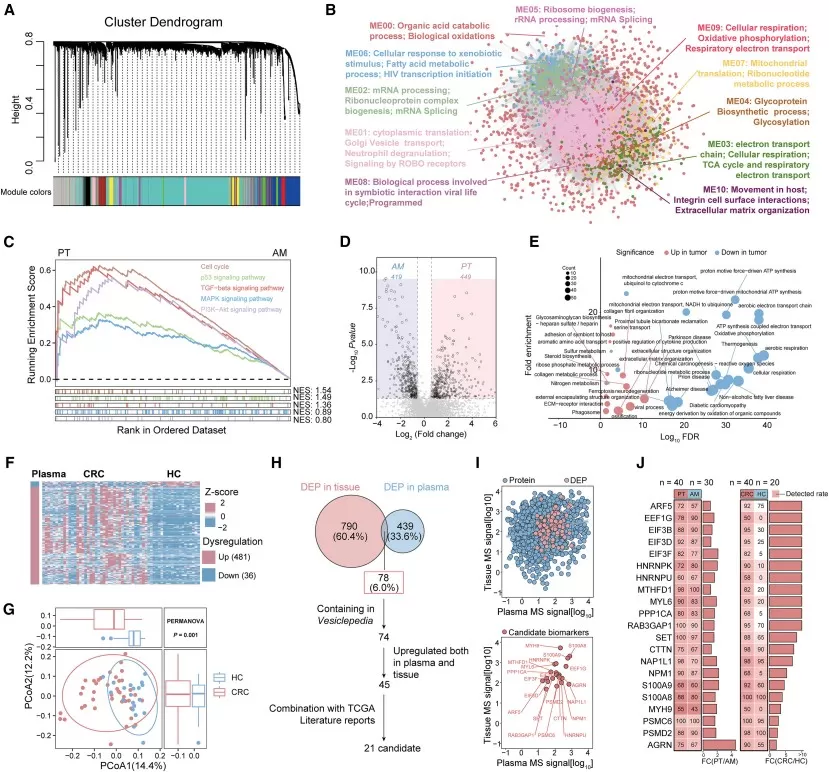
Figure 2. Integration of tissue EV and plasma EV proteomics revealed candidate biomarkers for CRC diagnosis. Image reproduced from Wang et al., 2025, Cell Reports Medicine, 6, 102090.
4. THERAPEUTIC TARGET DISCOVERY WITH ONCOPROTEOMICS
Oncoproteomics is also valuable for therapeutic target discovery because many drug-relevant mechanisms operate at the protein level. Genomic data can identify mutations, amplifications, or deletions, but these alterations do not always indicate whether a pathway is active, whether a kinase is signaling, or whether a compensatory resistance network has emerged. Proteomic and phosphoproteomic profiling can help answer these questions by measuring protein abundance, phosphorylation, pathway activation, metabolic programs, immune-related signals, and drug-response-associated protein changes. This makes oncoproteomics particularly useful for studying biological questions such as why tumors with similar mutations respond differently to treatment, which pathways are activated in resistant tumors, and which protein-level vulnerabilities may be prioritized for functional testing.
A representative example is the breast cancer serum EV proteomics study by Xu et al. The researchers used DIA-based mass spectrometry to profile serum EVs from patients with breast cancer and healthy donors in a discovery cohort, followed by five independent validation cohorts. This workflow identified EV protein classifiers for breast cancer diagnosis and metastatic disease assessment, but its most important therapeutic implication came from the discovery of TALDO1 as an EV-associated marker of distant metastasis.
The study did not stop at biomarker association. The researchers performed in vitro and in vivo experiments to test whether TALDO1 contributed to breast cancer progression. TALDO1 knockdown reduced tumor growth and progression, while EV-associated TALDO1 promoted invasive and metastatic phenotypes. These results supported TALDO1 as a metastasis-associated candidate therapeutic target rather than only a circulating biomarker. To explore targetability, the authors further conducted high-throughput molecular docking and virtual screening of 271,380 small molecules, identifying AO-022 as a TALDO1 allosteric inhibitor. AO-022 inhibited breast cancer cell migration in vitro and reduced tumor progression in vivo, providing preclinical evidence for TALDO1-targeted intervention. This case demonstrates how oncoproteomics can generate therapeutic hypotheses, prioritize functional targets, and guide early-stage drug discovery. However, TALDO1 and AO-022 should still be described as preclinical findings that require further validation before clinical application (Xu et al., 2024).
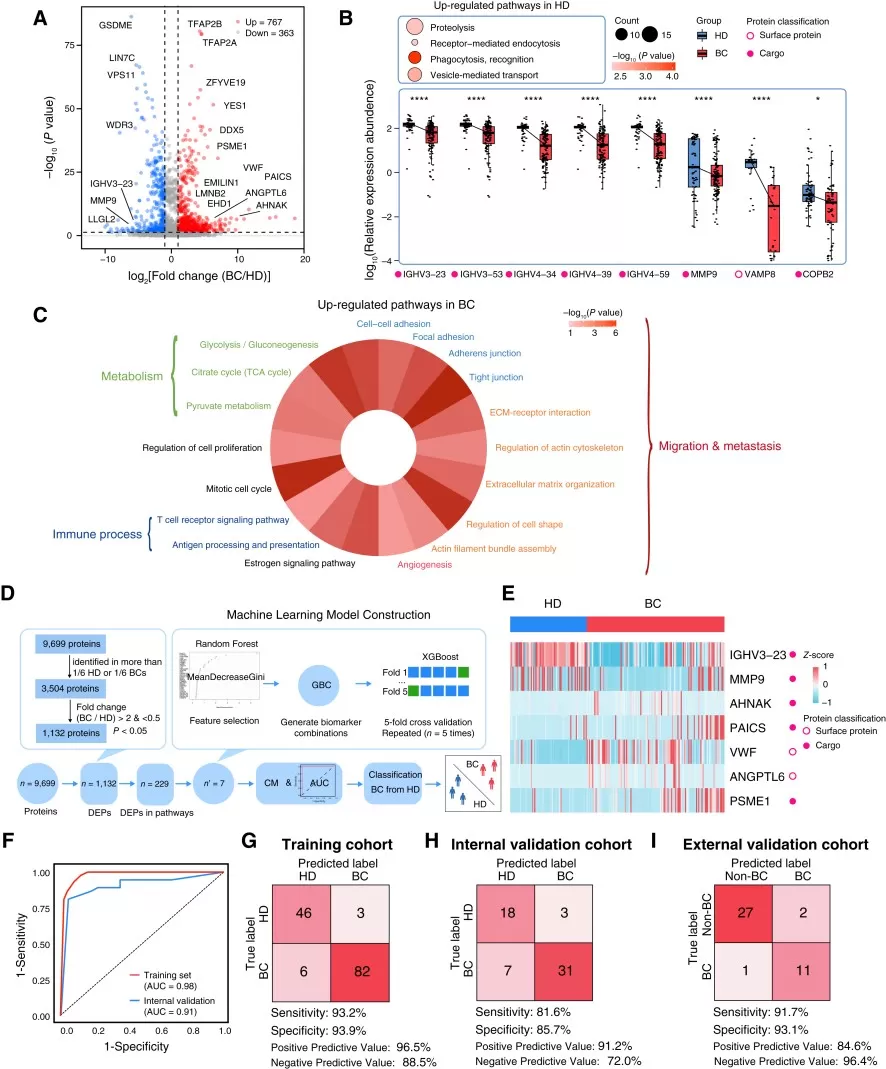
Figure 3. Proteomics features of breast cancer– and HD-derived EVs. Image reproduced from Xu et al., 2024, Cancer Research, 84(19), 3267–3285.
5. PRACTICAL CONSIDERATIONS FOR DESIGNING AN ONCOPROTEOMICS STUDY
A successful oncoproteomics study depends not only on the mass spectrometry platform, but also on how well the biological question, sample strategy, analytical workflow, and validation plan are aligned. Because cancer proteomics can be used for different purposes—from biomarker discovery to pathway analysis and therapeutic target prioritization—early study design decisions strongly influence the interpretability of the final results. The following considerations can help researchers build a more coherent discovery-to-validation workflow.
5.1 Define the Research Question
A well-designed oncoproteomics study should begin with a clear biological or translational question. Projects focused on diagnostic biomarker discovery, recurrence prediction, treatment resistance, pathway activation, or therapeutic target prioritization require different sample designs and analytical strategies. Defining the intended outcome at the beginning helps determine whether the study should emphasize broad protein discovery, PTM profiling, targeted validation, or multi-omics integration.
5.2 Select Appropriate Samples and Cohorts
Sample type strongly influences what biological questions can be answered. Fresh-frozen tumor tissue is often suitable for discovery-scale proteomics and PTM analysis, while FFPE tissue may be useful when clinically annotated archives are available. Plasma, serum, urine, and extracellular vesicles are valuable for liquid biopsy-oriented biomarker research, but they require careful control of pre-analytical variability and tissue-of-origin ambiguity. Cell lines, organoids, xenografts, and patient-derived models are more suitable for mechanistic studies of pathway activity, drug response, and resistance mechanisms. Cohort design should also consider tumor subtype, stage, treatment status, matched controls, adjacent tissue availability, and clinical endpoints.
5.3 Match the Analytical Workflow to the Objective
Global DIA quantitative proteomics is appropriate for unbiased discovery of protein abundance changes, dysregulated pathways, molecular subtypes, and candidate biomarkers. PTM-focused workflows, such as phosphoproteomics or glycoproteomics, are better suited for studying kinase signaling, cancer-associated glycoforms, or functional protein states. For validation, targeted approaches such as parallel reaction monitoring (PRM), selected reaction monitoring, targeted PTM assays, immunoassays, or orthogonal molecular readouts may provide more focused and reproducible evidence than repeating discovery proteomics alone.
5.4 Plan for Validation and Interpretation
Discovery-stage oncoproteomics results should be treated as candidate findings rather than immediate clinical conclusions. Reliable interpretation depends on adequate biological replication, randomized sample processing, balanced batches, quality-control samples, and predefined comparison groups. Independent cohorts, targeted quantitative assays, functional perturbation experiments, pharmacological testing, and integration with genomics, transcriptomics, metabolomics, histopathology, or clinical outcome data can strengthen the discovery-to-validation workflow and improve biological relevance.
6. CLINICAL TRANSLATION CHALLENGES AND FUTURE DIRECTIONS IN ONCOPROTEOMICS
Oncoproteomics provides valuable functional information for cancer research, but discovery results do not automatically translate into clinical biomarkers or therapeutic decisions. Several technical and biological factors can affect data quality, including tumor heterogeneity, stromal contamination, necrotic regions, ischemia time, sample storage conditions, limited cohort size, batch effects, and the difficulty of detecting low-abundance proteins. Body-fluid proteomics adds further complexity because circulating proteins can be influenced by inflammation, organ damage, medication, metabolic status, and other non-cancer conditions.
These limitations can be reduced through careful study design and standardized workflows. For tissue-based studies, strategies such as matched adjacent controls, histopathological review, tumor purity assessment, randomized sample processing, quality-control samples, and adequate biological replication can improve interpretability. For plasma, serum, urine, or extracellular vesicle studies, strict pre-analytical control, independent validation cohorts, and orthogonal assays are especially important. In PTM proteomics, enrichment efficiency, site localization confidence, and quantitative reproducibility should also be assessed before biological conclusions are drawn.
Clinical translation requires a higher evidentiary standard than discovery research. A candidate biomarker must demonstrate analytical reproducibility, biological relevance, independent validation, and clinically meaningful performance across relevant patient populations. Similarly, a candidate therapeutic target must be supported by functional experiments, pharmacological perturbation, disease-model evidence, and eventually clinical data. Therefore, oncoproteomics should be positioned as a high-value discovery and mechanistic research strategy, rather than a stand-alone guarantee of clinical diagnosis or treatment selection (Hristova and Chan, 2019; Haga et al., 2023).
Looking forward, the clinical value of oncoproteomics will depend on stronger integration between discovery-scale profiling and targeted validation. Improvements in DIA proteomics, targeted MS assays, single-cell and spatial proteomics, standardized sample handling, and multi-omics data integration are making protein-level evidence more reproducible and biologically interpretable. As these methods mature, oncoproteomics is expected to play an increasingly important role in prioritizing biomarker candidates, refining cancer subtypes, and generating testable therapeutic hypotheses for precision oncology research.
7. FAQ
7.1 Which sample types are suitable for oncoproteomics?
Oncoproteomics can be performed on tumor tissues, matched adjacent tissues, plasma, serum, urine, extracellular vesicles, cell models, organoids, xenografts, and other cancer-related samples. The optimal sample type depends on whether the study focuses on biomarker discovery, pathway activity, drug response, or therapeutic target prioritization.
7.2 Is serum or plasma proteomics suitable for cancer biomarker discovery?
Yes, but study design is critical. Serum and plasma are useful for non-invasive biomarker research, especially when combined with extracellular vesicle enrichment or targeted validation. However, circulating proteins can be affected by inflammation, organ damage, medication, and other non-cancer factors, so independent validation and appropriate controls are essential.
7.3 When should researchers choose global proteomics, phosphoproteomics, or glycoproteomics?
The choice depends on the biological question. Global proteomics is suitable when the goal is to broadly profile protein abundance, compare tumor and control groups, identify molecular subtypes, or discover candidate biomarkers. Phosphoproteomics is more appropriate when the study focuses on kinase signaling, pathway activation, drug response, or resistance mechanisms. Glycoproteomics should be considered when cancer-associated glycosylation, cell-surface proteins, secreted proteins, or disease-specific glycoforms are central to the hypothesis. In many oncoproteomics projects, these approaches can also be combined to connect protein abundance with functional protein states.
How MetwareBio Can Support Oncoproteomics Research
As oncoproteomics becomes increasingly important in cancer biomarker discovery, therapeutic target identification, and drug-response studies, researchers need workflows that connect reliable proteomic measurement with clear biological interpretation.
MetwareBio provides full-stack, mass spectrometry-based proteomics services to support projects from experimental design to data analysis. Depending on the research objective, MetwareBio can support global proteome profiling, blood proteomics, diverse PTM proteomics analyses (including phosphoproteomics, glycoproteomics, ubiquitination, acetylation, and more), protein complex analysis, and multi-omics integration. These workflows can be adapted to tumor tissues, body fluids, extracellular vesicles, cell models, organoids, xenografts, and other cancer-related samples.
If you are interested in oncoproteomics or cancer proteomics services, please do not hesitate to contact us.
Contact UsRead More: Cancer Proteomics and Multi-Omics Approaches
Oncoproteomics integrates with a broad range of proteomics technologies and analytical strategies. The articles below cover complementary methods—from PTM analysis and clinical proteomics to multi-omics integration—that expand the toolkit for cancer biomarker discovery and therapeutic target identification.
Explore how clinical proteomics bridges discovery-stage protein profiling with real-world diagnostic and therapeutic applications. This article addresses the key bottlenecks—sample variability, cohort design, and validation standards—that oncoproteomics studies must overcome on the path to clinical translation.
Post-translational modifications are central to oncoproteomics—from phosphorylation in kinase signaling to glycosylation in tumor cell-surface biology. This article covers the computational workflow for PTM identification, including site localization scoring, FDR control, and interpretation of modification-specific results.
A comprehensive guide to processing and interpreting proteomics data generated by LC-MS/MS—the core technology behind most oncoproteomics workflows. Covers database searching, protein inference, quantification strategies, and quality control steps from raw data to biological conclusions.
Oncoproteomics gains additional resolving power when integrated with metabolomics. This article explains how joint protein–metabolite profiling can reveal metabolic rewiring in tumors, identify biomarker panels with complementary specificity, and support multi-dimensional therapeutic target discovery in cancer research.
Data-independent acquisition (DIA) is increasingly the preferred strategy for large-scale oncoproteomics discovery cohorts due to its reproducibility and broad proteome coverage. Learn how DIA-MS enables deep, unbiased protein quantification across tumor samples, body fluids, and extracellular vesicles.
Cancer biology is shaped by interactions across the genome, transcriptome, proteome, and metabolome. This overview of MetwareBio's multi-omics capabilities explains how integrating proteomics with genomic, transcriptomic, and metabolomic data creates a more complete picture of tumor mechanisms and strengthens the evidence base for biomarker and therapeutic target validation.
References
- Aebersold, R., & Mann, M. (2016). Mass-spectrometric exploration of proteome structure and function. Nature, 537(7620), 347–355. https://doi.org/10.1038/nature19949
- Dutta, S., Ghosh, S., Mishra, A., et al. (2023). Oncoproteomics: insight into current proteomic technologies in cancer biomarker discovery and treatment. Journal of Proteins and Proteomics, 14, 1–24. https://doi.org/10.1007/s42485-022-00100-6
- Gatto, L., Aebersold, R., Cox, J., Demichev, V., Derks, J., Emmott, E., et al. (2023). Initial recommendations for performing, benchmarking and reporting single-cell proteomics experiments. Nature Methods, 20, 375–386. https://doi.org/10.1038/s41592-023-01785-3
- Haga, Y., Minegishi, Y., & Ueda, K. (2023). Frontiers in mass spectrometry-based clinical proteomics for cancer diagnosis and treatment. Cancer Science, 114(5), 1783–1791. https://doi.org/10.1111/cas.15731
- Hristova, V. A., & Chan, D. W. (2019). Cancer biomarker discovery and translation: proteomics and beyond. Expert Review of Proteomics, 16(2), 93–103. https://doi.org/10.1080/14789450.2019.1579613
- Mani, D. R., Krug, K., Zhang, B., et al. (2022). Cancer proteogenomics: current impact and future prospects. Nature Reviews Cancer, 22(5), 298–313. https://doi.org/10.1038/s41568-022-00446-5
- Pinho, S. S., & Reis, C. A. (2015). Glycosylation in cancer: mechanisms and clinical implications. Nature Reviews Cancer, 15(9), 540–555. https://doi.org/10.1038/nrc3982
- Wang, J., Gu, C.-Z., Wang, P.-X., et al. (2025). Integrative proteomic profiling of tumor and plasma extracellular vesicles identifies a diagnostic biomarker panel for colorectal cancer. Cell Reports Medicine, 6, 102090. https://doi.org/10.1016/j.xcrm.2025.102090
- Xu, G., Huang, R., Wumaier, R., et al. (2024). Proteomic profiling of serum extracellular vesicles identifies diagnostic signatures and therapeutic targets in breast cancer. Cancer Research, 84(19), 3267–3285. https://doi.org/10.1158/0008-5472.CAN-23-3998
