
Quantitative proteomics is widely used to identify differentially expressed proteins across biological conditions, disease models, treatment groups, and clinical cohorts. Discovery workflows such as label-free quantification and TMT-based proteomics, combined with data-dependent acquisition or data-independent acquisition, can generate comprehensive protein profiles and reveal candidate biomarkers, regulatory proteins, and pathway-level changes. However, discovery proteomics is usually the starting point for biological interpretation rather than a final experimental endpoint. Once candidate proteins have been prioritized, their abundance changes often need to be confirmed using independent or complementary validation methods. This step is particularly important when proteomics findings will be used to support biomarker development, pathway interpretation, or mechanistic follow-up. This article summarizes major experimental validation strategies for quantitative proteomics results, including targeted mass spectrometry, antibody-based assays, and functional or mechanistic follow-up experiments. It also discusses how these approaches can be combined into a staged biomarker discovery and validation workflow, helping researchers select appropriate validation strategies based on different research goals.
1. WHY QUANTITATIVE PROTEOMICS RESULTS NEED VALIDATION
Quantitative proteomics can generate strong candidate lists, but the transition from a discovery result to a biological conclusion requires careful confirmation. Even when sample quality control and statistical analysis are appropriate, protein-level changes may still be affected by sample heterogeneity, peptide detectability, protein inference, missing values, batch effects, and the biological variability of the study system. Validation helps determine whether selected proteins show reproducible changes under the relevant experimental or clinical conditions.
The goal of validation is usually not to repeat the entire discovery dataset. Instead, validation focuses on a smaller set of candidate proteins selected for their statistical strength, biological relevance, pathway position, biomarker potential, or feasibility for downstream assays. A validated result does not necessarily need to reproduce the exact fold change observed in discovery proteomics, because different methods measure protein signals through different analytical principles. A useful validation result should support the same direction of change, show reasonable sample-level consistency, and fit the biological context of the study.
2. MASS SPECTROMETRY-BASED TARGETED PROTEOMICS VALIDATION
Targeted mass spectrometry is often one of the most direct approaches for validating quantitative proteomics results because it remains within the LC-MS/MS analytical framework. It is especially useful when multiple candidate proteins need to be confirmed or when high-quality antibodies are not available.
2.1 PRM and MRM: Technical Principles
Targeted proteomics measures predefined peptides that represent selected proteins. The two commonly used strategies are parallel reaction monitoring (PRM) and multiple reaction monitoring (MRM), also known as selected reaction monitoring (SRM). MRM is typically performed on triple quadrupole instruments and monitors predefined precursor-to-fragment ion transitions. PRM is commonly performed on high-resolution mass spectrometers, where selected precursor ions are isolated and multiple fragment ions are recorded for peptide confirmation and quantification (Peterson et al., 2012).
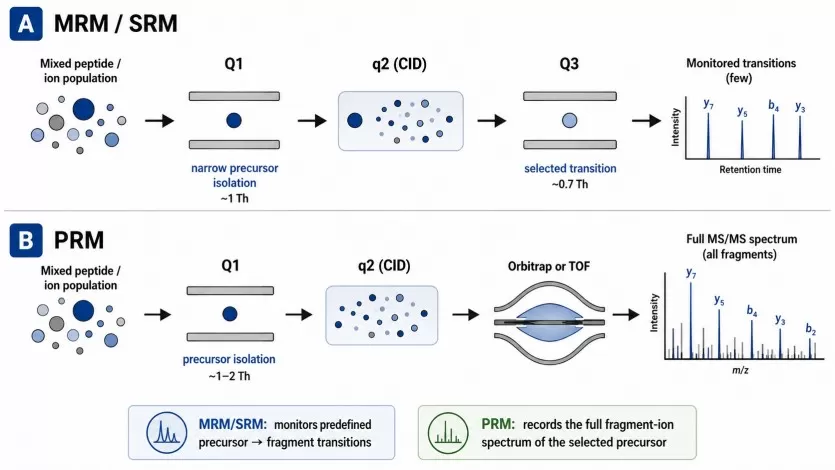
Figure 1. Schematic representation of SRM (A) and PRM (B) as performed on QqQ and QqOrbi (or QqTOF) instrumentation, respectively. Image adapted from Peterson et al., 2012, Molecular & Cellular Proteomics, 11(11), 1475–1488.
Compared with antibody-based assays, PRM/MRM does not depend on antibody availability or antibody specificity. This makes targeted MS valuable for proteins with poor antibody coverage, homologous protein families, isoform-related ambiguity, or multi-protein biomarker panels. In addition, targeted MS can quantify several proteins in the same analytical run. Software ecosystems such as Skyline support quantitative mass spectrometry workflows, including SRM/MRM, PRM, DIA/SWATH, and related targeted data analysis tasks (Pino et al., 2020). Standardized peptide and proteoform annotation frameworks also help improve consistency in reporting peptide-level measurements and modified sequences (LeDuc et al., 2022).
2.2 PRM/MRM Validation Workflow
A typical targeted proteomics validation workflow begins with candidate protein prioritization from the discovery dataset. Candidate selection usually considers fold change, statistical significance, peptide evidence, biological relevance, pathway involvement, and technical detectability. The most suitable candidates are not always the proteins with the largest fold changes; they are often proteins that combine robust quantitative evidence with a clear biological question.
The next step is proteotypic peptide selection. Ideal peptides should be unique to the target protein, reproducibly detected, compatible with LC-MS/MS analysis, and free from problematic sequence features when possible. Peptides with frequent missed cleavages, unstable modifications, severe interference, or shared sequences may reduce confidence and should be evaluated carefully.
After peptide selection, a targeted acquisition method is developed. For MRM, optimized transitions are monitored. For PRM, selected precursor ions are fragmented and quantified using fragment ion chromatograms. Stable isotope-labeled peptides or other internal standards can improve quantitative robustness when higher precision is required. Validation samples are then analyzed, preferably using independent biological samples or expanded cohorts when reproducibility is the central goal. Results are interpreted by comparing abundance direction, quantitative trend, statistical significance, and sample-level consistency with the original discovery proteomics results.
3. ANTIBODY-BASED Biochemical Validation
3.1 Western Blot for Selected Protein Confirmation
Western blot is commonly used to validate one or a few candidate proteins in cell lysates, tissue homogenates, or other extracted protein samples. It provides visual information about apparent molecular weight, band pattern, and relative protein abundance (Mahmood and Yang, 2012). This makes it useful when the expected protein size is known or when the study needs to distinguish full-length proteins from major cleavage products or large isoform differences.
For proteomics validation, Western blot is most appropriate when a reliable antibody is available and the number of targets is limited. It can support the abundance trend observed in discovery proteomics, especially when the same direction of change is observed across biological replicates. However, Western blot should not be treated as automatically definitive. Non-specific bands, weak signal, cross-reactivity, saturation of signal intensity, inconsistent transfer efficiency, and unsuitable loading controls can all affect interpretation. Appropriate antibody validation, linear exposure range, biological replicates, and consistent normalization are essential.
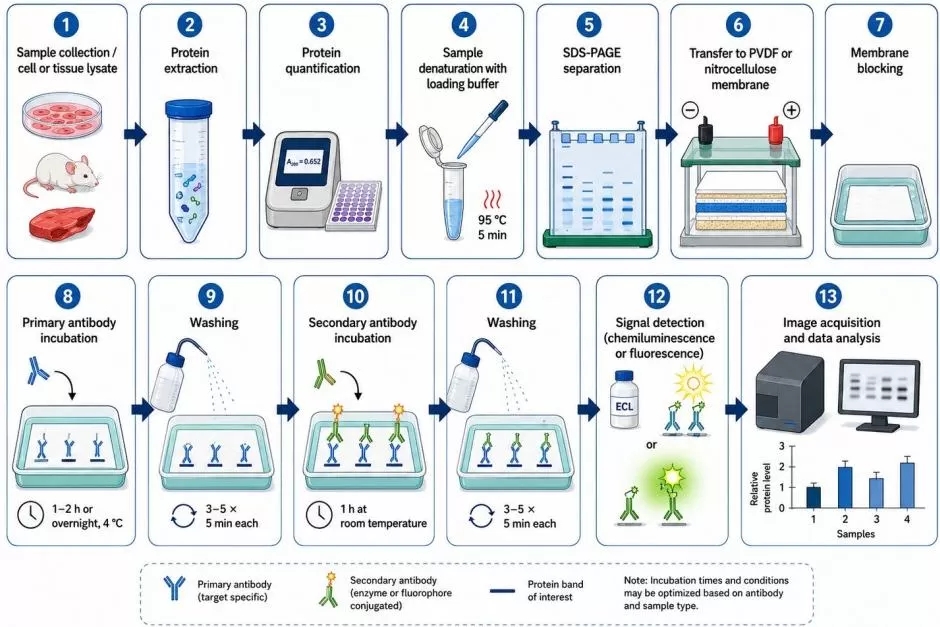
Figure 2. The Experimental Workflow of Western Blot.
3.2 ELISA for Soluble Proteins and Biomarkers
ELISA is useful for validating soluble or secreted proteins in sample types such as plasma, serum, cerebrospinal fluid, urine, cell culture supernatant, and tissue homogenate. It is often applied to cytokines, chemokines, hormones, growth factors, inflammatory mediators, and candidate biomarkers. When a validated kit is available, ELISA can provide concentration-based measurements and may be more scalable than Western blot for larger sample cohorts (Aydin, 2015).
This method is particularly relevant for translational or biomarker-oriented proteomics studies. For example, if discovery proteomics identifies a secreted protein that differs between disease and control groups, ELISA may help evaluate whether the same trend is detectable in additional samples. The major considerations include antibody pair specificity, calibration curve quality, dilution linearity, dynamic range, matrix compatibility, and sample handling consistency. Plasma and serum samples may contain interfering components, and inappropriate dilution can obscure true differences. Rigorous assay controls are therefore required for reliable interpretation. (Learn more at: ELISA vs. Western Blot)
3.3 IHC for Tissue-Level Localization
Immunohistochemistry (IHC) is valuable when tissue context is important. Bulk proteomics can indicate that a protein is increased or decreased in a tissue sample, but it usually cannot determine whether the change occurs in tumor cells, stromal cells, immune cells, epithelial compartments, necrotic regions, or specific anatomical structures. IHC can add this spatial layer by localizing candidate proteins in formalin-fixed paraffin-embedded or frozen tissue sections.
IHC is commonly used in cancer research, pathology-related studies, inflammatory disease, organ injury models, and tissue biomarker evaluation. It can help connect proteomics findings with histological features, disease regions, or cell-type distribution. Large antibody-based resources, including the Human Protein Atlas, illustrate how tissue profiling and antibody validation can support the interpretation of protein expression patterns across tissues and disease contexts (Sivertsson et al., 2020). However, IHC results depend on antibody specificity, antigen retrieval, staining optimization, scoring strategy, and tissue heterogeneity. Positive controls, negative controls, standardized staining conditions, and reproducible image analysis are important for confidence.
3.4 IF for Cellular and Subcellular Localization
Immunofluorescence (IF) is suitable when the validation question involves cellular localization, subcellular distribution, or co-localization with markers. IF can help determine whether a protein is enriched in the nucleus, cytoplasm, plasma membrane, mitochondria, endoplasmic reticulum, cytoskeleton, or other cellular compartments. It can also be combined with cell-type markers, organelle markers, or pathway markers to evaluate spatial relationships.
For proteomics validation, IF is especially useful when total abundance alone is insufficient. A treatment may not only increase or decrease a protein, but may also alter where the protein is located within the cell. Such redistribution can be biologically meaningful for transcription factors, signaling molecules, membrane proteins, and organelle-associated proteins. Large-scale subcellular proteome mapping has demonstrated the value of immunofluorescence imaging for assigning proteins to cellular compartments and interpreting localization-dependent biology (Thul et al., 2017). IF interpretation requires careful control of autofluorescence, non-specific staining, spectral overlap, antibody cross-reactivity, and imaging settings. Quantitative IF should use consistent acquisition parameters and objective image analysis when possible.
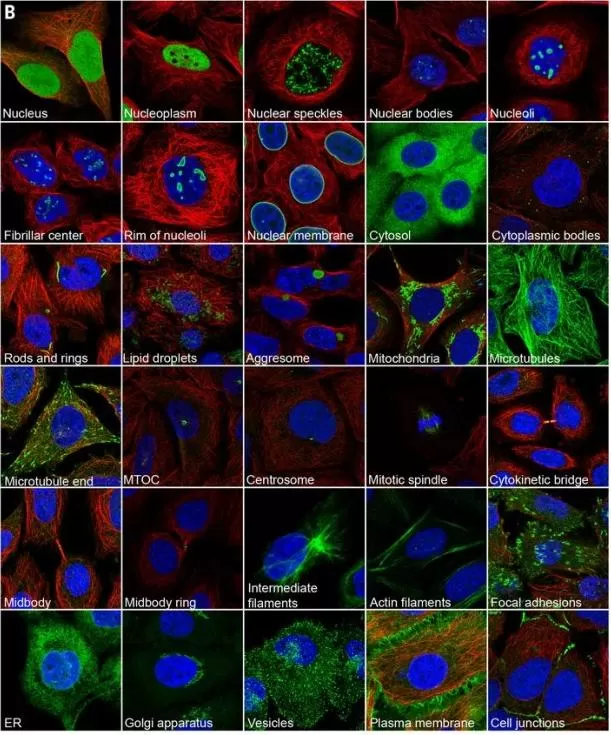
Figure 3. Subcellular structures annotated in the Cell Atlas by immunofluorescence (IF) microscopy. Examples of proteins (green) localizing to each annotated structure in the representative set of human cell lines used in the Cell Atlas. Microtubules are marked with an antibody against tubulin (red); the nucleus is counterstained with DAPI (blue). Image reproduced from Thul et al., 2017, Science, 356, eaal3321.
4. FUNCTIONAL AND MECHANISTIC VALIDATION AFTER PROTEIN-LEVEL CONFIRMATION
Protein-level validation addresses whether the abundance change observed in discovery proteomics is reproducible at the protein level. Functional and mechanistic validation addresses a different question: whether the validated candidate protein is likely to contribute to the phenotype, pathway, or biological process observed in the study. These experiments are typically performed after abundance changes have been confirmed by protein-level methods.
4.1 Perturbation Followed by Phenotype Readouts
Gene or protein perturbation experiments test whether changing the level or activity of a candidate protein affects a measurable biological outcome. Common strategies include siRNA knockdown, shRNA knockdown, CRISPR knockout, CRISPR interference, CRISPR activation, overexpression, and rescue experiments. If a protein is upregulated in discovery proteomics and confirmed by validation, knockdown or knockout may help test whether reducing its expression weakens the phenotype of interest. If a protein is downregulated, overexpression or rescue may help evaluate whether restoring its level modifies the phenotype.
The phenotype readout should match the biological context. In cancer studies, appropriate readouts may include proliferation, colony formation, EdU incorporation, apoptosis, cell cycle progression, wound healing, invasion, and migration. In metabolic research, readouts may include ATP production, glucose uptake, lactate release, mitochondrial membrane potential, oxygen consumption rate, extracellular acidification rate, and reactive oxygen species levels. In immune or inflammatory studies, cytokine release, immune cell activation, macrophage polarization, or flow cytometry-based marker analysis may be more appropriate. These experiments do not directly validate the proteomics quantification; they test whether a validated protein is functionally related to the observed phenotype.
4.2 Pathway and Enzyme Activity Assays
Proteomics results often suggest pathway-level changes rather than isolated protein effects. Pathway activity assays can help evaluate whether an inferred signaling, metabolic, or stress-response pathway is functionally altered. Depending on the hypothesis, these assays may involve phosphorylation analysis, downstream marker detection, reporter assays, enzyme activity measurements, targeted metabolite assays, or cellular bioenergetic readouts.
For example, if proteomics suggests changes in PI3K-AKT, MAPK, NF-kB, apoptosis, autophagy, glycolysis, oxidative phosphorylation, extracellular matrix remodeling, or inflammatory signaling, pathway-level validation may focus on key phosphorylated proteins, enzymatic outputs, transcriptional reporters, or pathway-associated metabolites. Curated pathway resources such as Reactome can support pathway interpretation, while protein association resources such as STRING can help identify functional relationships among candidate proteins (Gillespie et al., 2022; Szklarczyk et al., 2023). These tools are useful for hypothesis generation, but experimental readouts are still needed to support pathway activity in the specific biological model.
4.3 Interaction and Regulatory Mechanism Assays
When a candidate protein is suspected to regulate other proteins or participate in a protein complex, interaction assays may be needed. Common approaches include co-immunoprecipitation, pull-down assays, proximity ligation assays, and immunoprecipitation followed by mass spectrometry. These methods can help determine whether the candidate protein physically or functionally associates with predicted partners.
For transcriptional regulators, mechanistic validation may involve ChIP-qPCR, ChIP-seq, luciferase reporter assays, or downstream gene expression analysis. For enzymes, activity assays may be more informative than abundance measurements alone, because protein concentration and catalytic activity are not always equivalent. For signaling proteins, phosphorylation status, localization, and complex formation may provide more mechanistic insight than total protein abundance. These experiments should be positioned as mechanistic follow-up rather than first-line validation of quantitative proteomics results.
5. HOW TO CHOOSE THE RIGHT PROTEOMICS VALIDATION STRATEGY
The appropriate validation strategy depends on the validation goal, protein class, sample matrix, available reagents, and downstream research plan. For direct confirmation of multiple protein abundance changes, targeted mass spectrometry is often a strong option because it provides peptide-level, antibody-independent quantification. For a small number of proteins with reliable antibodies, Western blot may be appropriate. For soluble or secreted biomarker candidates, ELISA may be more practical. For spatial interpretation, IHC and IF can provide tissue, cellular, or subcellular context that bulk proteomics cannot capture. Functional and mechanistic experiments should be selected only after the biological question is clear. They are not substitutes for protein-level validation. Instead, they help determine whether a confirmed protein abundance change has functional relevance, pathway involvement, or mechanistic importance.
Comparison of Experimental Validation Methods for Quantitative Proteomics Results
| Validation Goal | Suitable Method | Best Use Case | Main Strength | Key Considerations |
|---|---|---|---|---|
| Confirm multiple candidate proteins at the protein level | PRM/MRM targeted proteomics | Discovery proteomics follow-up; multi-protein panels; proteins without reliable antibodies | Direct MS-based quantification; multiplexed; antibody-independent | Requires peptide selection, method development, and appropriate internal controls |
| Validate one or a few proteins in extracted samples | Western blot | Cell lysates, tissue homogenates, or protein extracts with reliable antibodies | Visual band pattern and relative abundance information | Semi-quantitative; affected by antibody specificity, loading controls, and exposure range |
| Measure soluble or secreted proteins | ELISA | Plasma, serum, CSF, urine, cell supernatant, or tissue homogenate | Sensitive and scalable for larger cohorts when a validated kit is available | Matrix effects, dynamic range, dilution linearity, and kit quality require evaluation |
| Confirm tissue-level localization | IHC | FFPE or frozen tissue sections; pathology-related interpretation | Shows tissue distribution and regional expression patterns | Antibody specificity, antigen retrieval, scoring strategy, and tissue heterogeneity are critical |
| Assess cellular or subcellular localization | IF | Cell models or tissue sections requiring co-localization analysis | Supports localization, redistribution, and cell-context interpretation | Requires imaging controls, spectral separation, and consistent acquisition settings |
| Evaluate transcript-level consistency | qPCR | Testing whether mRNA trends are consistent with protein changes | Useful for interpreting transcriptional regulation | Does not directly validate protein abundance |
| Test functional relevance | Perturbation plus phenotype readouts | Determining whether a validated protein affects a phenotype | Supports functional interpretation | Requires model-specific controls and careful phenotype selection |
| Investigate pathway or mechanism | Pathway assays, enzyme activity, Co-IP, pull-down, reporter assays | Mechanistic follow-up after protein-level validation | Links candidates to pathways, interactions, or regulatory mechanisms | Should complement, not replace, abundance validation |
6. PROTEOMICS-BASED BIOMARKER DISCOVERY AND VALIDATION WORKFLOW
In biomarker-oriented proteomics studies, validation is rarely completed by a single method. A more reliable strategy is to build evidence step by step, moving from discovery proteomics to targeted verification, orthogonal validation, cohort-level testing, and, when needed, functional follow-up.
A colorectal cancer EV proteomics study provides a clear example of this staged approach. The researchers first integrated tissue EV and plasma EV proteomes to identify candidate diagnostic proteins. Selected candidates were then verified by PRM-MS, which provided targeted peptide-level confirmation before the study moved into ELISA-based validation in larger clinical cohorts. The final biomarker model was further evaluated in internal and external cohorts, showing how discovery proteomics, targeted mass spectrometry, antibody-based assays, and cohort validation can be combined to support molecular marker development (Wang et al., 2025).
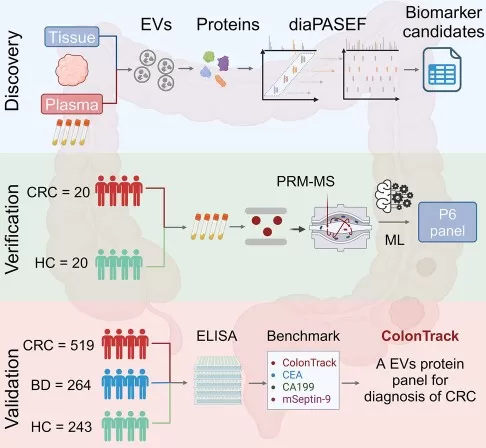
Figure 4. Biomarker Discovery and Validation Workflow. Image reproduced from Wang et al., 2025, Cell Reports Medicine, 6, 102090.
A breast cancer serum EV proteomics study illustrates a complementary strategy. DIA-based proteomics was used to identify EV protein signatures associated with breast cancer diagnosis and metastasis. The findings were tested across multiple independent cohorts, and TALDO1 was further evaluated by tissue-level IHC and functional experiments. This design connected the circulating EV proteomics signal with tissue expression patterns and biological relevance, strengthening the interpretation of TALDO1 as both a metastasis-associated biomarker and a potential functional target (Xu et al., 2024).
Together, these two studies demonstrate that molecular marker identification after quantitative proteomics is best viewed as a staged evidence-building process. Discovery proteomics identifies candidates. Targeted MS verifies selected proteins at the peptide level. ELISA, Western blot, IHC, or IF provide orthogonal validation depending on the sample type and biological question. Large independent cohorts test reproducibility and diagnostic performance. Functional and mechanistic assays are added when the candidate protein may influence the phenotype or pathway under investigation. A practical validation strategy may therefore follow this sequence:
- Use discovery proteomics to identify candidate proteins with robust quantitative changes.
- Prioritize candidates based on statistical strength, biological relevance, detectability, sample type, and intended application.
- Use PRM/MRM targeted proteomics to verify selected candidates in a focused sample set.
- Use antibody-based assays such as ELISA, Western blot, IHC, or IF to provide orthogonal validation or spatial context.
- Evaluate diagnostic or prognostic performance in independent and preferably larger cohorts.
- Add perturbation, pathway, or interaction assays when the candidate biomarker is also hypothesized to have functional or mechanistic relevance.
This staged approach helps distinguish a differentially expressed protein from a biologically and clinically meaningful molecular marker. It also reduces the risk of overinterpreting discovery proteomics results before reproducibility, assay feasibility, and biological context have been adequately evaluated.
How MetwareBio Supports Proteomics Follow-Up and Multi-Omics Research
After candidate proteins are identified, the next step is to strengthen biological interpretation with the right analytical platform. MetwareBio supports research projects with mass spectrometry-based proteomics, Olink, SomaScan, and integrated multi-omics analysis. Contact us to discuss your sample type, research goals, and how proteomics data can be connected with broader molecular evidence.
Contact UsRead More: Proteomics Validation, Targeted Quantification, and Biomarker Workflows
Explore these related articles to deepen your understanding of targeted proteomics methods, validation assays, and biomarker discovery workflows — from PRM/MRM principles to quality control practices and multi-cohort biomarker evaluation.
A detailed guide to PRM as a targeted proteomics validation tool. Learn how PRM works on high-resolution instruments, why it outperforms antibody-based assays for multi-protein confirmation, and how to design a PRM experiment after discovery proteomics identifies your candidate proteins.
Understand the technical differences between PRM (high-resolution) and MRM (triple quadrupole) for targeted proteomics validation. This comparison helps you choose the right method based on your instrument availability, panel size, and required quantification accuracy when confirming proteomics hits.
A comprehensive practical guide to Western blot as an orthogonal proteomics validation method. Covers antibody selection, loading controls, exposure optimization, and common pitfalls — providing the technical foundation needed to interpret Western blot results alongside discovery proteomics data.
Compare ELISA and Western blot side by side for proteomics result validation. This article helps researchers decide which immunoassay best fits their validation goal — whether confirming soluble biomarker candidates in plasma and serum (ELISA) or checking protein expression in cell or tissue lysates (Western blot).
A step-by-step walkthrough of the complete proteomics biomarker discovery pipeline — from sample preparation and quantitative data acquisition through statistical analysis, candidate prioritization, and staged validation. Directly complementary to the multi-method validation framework described in this article.
Before validation can begin, discovery proteomics data must meet quality thresholds. This guide covers QC checkpoints at every stage — from sample preparation and LC-MS/MS acquisition to missing value assessment and batch effect monitoring — ensuring your candidate protein list is built on solid, reproducible data.
References
- Gillespie, M., Jassal, B., Stephan, R., Milacic, M., Rothfels, K., Senff-Ribeiro, A., Griss, J., Sevilla, C., Matthews, L., Gong, C., Deng, C., Varusai, T., Ragueneau, E., Haider, Y., May, B., Shamovsky, V., Weiser, J., Brunson, T., Sanati, N., Beckman, L., … D'Eustachio, P. (2022). The reactome pathway knowledgebase 2022. Nucleic acids research, 50(D1), D687–D692. https://doi.org/10.1093/nar/gkab1028
- LeDuc, R. D., Deutsch, E. W., Binz, P. A., Fellers, R. T., Cesnik, A. J., Klein, J. A., Van Den Bossche, T., Gabriels, R., Yalavarthi, A., Perez-Riverol, Y., Carver, J., Bittremieux, W., Kawano, S., Pullman, B., Bandeira, N., Kelleher, N. L., Thomas, P. M., & Vizcaíno, J. A. (2022). Proteomics Standards Initiative's ProForma 2.0: Unifying the Encoding of Proteoforms and Peptidoforms. Journal of proteome research, 21(4), 1189–1195. https://doi.org/10.1021/acs.jproteome.1c00771
- Pino, L. K., Searle, B. C., Bollinger, J. G., Nunn, B., MacLean, B., & MacCoss, M. J. (2020). The Skyline ecosystem: Informatics for quantitative mass spectrometry proteomics. Mass spectrometry reviews, 39(3), 229–244. https://doi.org/10.1002/mas.21540
- Sivertsson, Å., Lindström, E., Oksvold, P., Katona, B., Hikmet, F., Vuu, J., Gustavsson, J., Sjöstedt, E., von Feilitzen, K., Kampf, C., Schwenk, J. M., Uhlén, M., & Lindskog, C. (2020). Enhanced Validation of Antibodies Enables the Discovery of Missing Proteins. Journal of proteome research, 19(12), 4766–4781. https://doi.org/10.1021/acs.jproteome.0c00486
- Szklarczyk, D., Kirsch, R., Koutrouli, M., Nastou, K., Mehryary, F., Hachilif, R., Gable, A. L., Fang, T., Doncheva, N. T., Pyysalo, S., Bork, P., Jensen, L. J., & von Mering, C. (2023). The STRING database in 2023: protein-protein association networks and functional enrichment analyses for any sequenced genome of interest. Nucleic acids research, 51(D1), D638–D646. https://doi.org/10.1093/nar/gkac1000
- Peterson, A. C., Russell, J. D., Bailey, D. J., Westphall, M. S., & Coon, J. J. (2012). Parallel reaction monitoring for high resolution and high mass accuracy quantitative, targeted proteomics. Molecular & Cellular Proteomics, 11(11), 1475–1488. https://doi.org/10.1074/mcp.O112.020131
- Mahmood, T., & Yang, P.-C. (2012). Western blot: Technique, theory, and trouble shooting. North American Journal of Medical Sciences, 4(9), 429–434. https://doi.org/10.4103/1947-2714.100998
- Aydin, S. (2015). A short history, principles, and types of ELISA, and our laboratory experience with peptide/protein analyses using ELISA. Peptides, 72, 4–15. https://doi.org/10.1016/j.peptides.2015.04.012
- Edfors, F., Hober, A., Linderbäck, K., Maddalo, G., Azimi, A., Sivertsson, Å., Tegel, H., Hober, S., Szigyarto, C. A.-K., Fagerberg, L., von Feilitzen, K., Oksvold, P., Lindskog, C., Forsström, B., Uhlén, M., & Pontén, F. (2018). Enhanced validation of antibodies for research applications. Nature Communications, 9, 4130. https://doi.org/10.1038/s41467-018-06642-y
- Thul, P. J., Åkesson, L., Wiking, M., Mahdessian, D., Geladaki, A., Ait Blal, H., Alm, T., Asplund, A., Björk, L., Breckels, L. M., Bäckström, A., Danielsson, F., Fagerberg, L., Fall, J., Gatto, L., Gnann, C., Hober, S., Hjelmare, M., Johansson, F., ... Lundberg, E. (2017). A subcellular map of the human proteome. Science, 356(6340), eaal3321. https://doi.org/10.1126/science.aal3321
- Xu, G., Huang, R., Wumaier, R., Lyu, J., Huang, M., Zhang, Y., Chen, Q., Liu, W., Tao, M., Li, J., Tao, Z., Yu, B., Xu, E., Wang, L., Yu, G., Gires, O., Zhou, L., Zhu, W., Ding, C., & Wang, H. (2024). Proteomic profiling of serum extracellular vesicles identifies diagnostic signatures and therapeutic targets in breast cancer. Cancer Research, 84(19), 3267–3285. https://doi.org/10.1158/0008-5472.CAN-23-3998
- Wang, J., Gu, C.-Z., Wang, P.-X., Xian, J.-R., Wang, H., Shang, A.-Q., Zhong, Y.-C., Zheng, W.-J., Cheng, J.-W., Yang, W.-J., Zhou, J., Fan, J., Guo, W., Yang, X.-R., & Lu, H.-J. (2025). Integrative proteomic profiling of tumor and plasma extracellular vesicles identifies a diagnostic biomarker panel for colorectal cancer. Cell Reports Medicine, 6, 102090. https://doi.org/10.1016/j.xcrm.2025.102090
