
Top-Down vs. Bottom-Up Proteomics: Unraveling the Secrets of Protein Analysis
Outline:
- Introduction to Proteomics
- Top-Down Proteomics: Analyzing Intact Proteins
- Workflow of Top-Down Proteomics
- Characteristics and Limitations of Top-Down Proteomics
- Bottom-Up Proteomics: Fragmenting Proteins for Analysis
- Workflow of Bottom-Up Proteomics: Detailed Breakdown
- Advantages and Limitations of Bottom-Up Proteomics
- Top-Down vs. Bottom-Up Proteomics: Comparative Summary
- Conclusion
Introduction to Proteomics
Proteomics, the large-scale study of proteins, plays a crucial role in understanding biological processes and disease mechanisms. It encompasses the identification, quantification, and characterization of proteins, providing insights into their functions and interactions within cells. Within the field of proteomics, two primary strategies based on mass spectrometry are commonly employed: Top-Down Proteomics and Bottom-Up Proteomics. These approaches differ significantly in their handling methods, application areas, and inherent advantages and disadvantages.
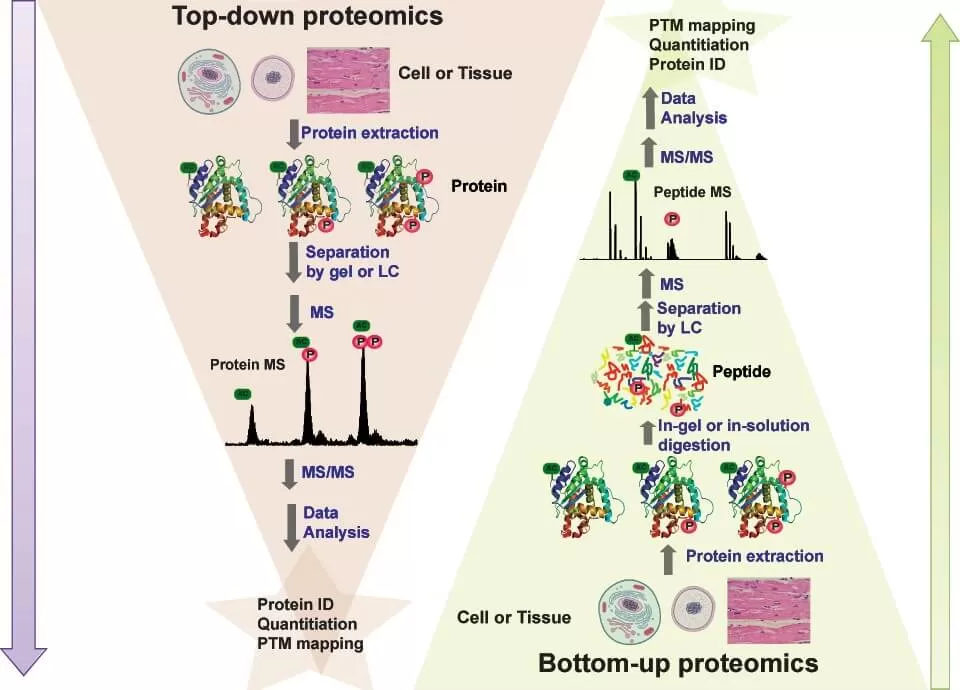
Top-Down Proteomics: Analyzing Intact Proteins
Top-Down Proteomics refers to the method of mass spectrometry analysis conducted directly on intact proteins without any enzymatic digestion or chemical treatment. Unlike traditional methods that fragment proteins into peptides, Top-Down Proteomics directly examines whole proteins, enabling comprehensive characterization of their proteoforms—distinct protein variants arising from genetic variations, alternative splicing, and post-translational modifications (PTMs). This global analysis is performed using high-resolution mass spectrometry techniques, such as Fourier-transform ion cyclotron resonance (FT-ICR) or Orbitrap, which fragment intact proteins to reveal their molecular details.
The primary advantages of Top-Down Proteomics include the ability to avoid false-positive identifications and the simultaneous detection of multiple post-translational modification sites. Additionally, it effectively quantifies and distinguishes different proteoforms, capturing the true information of proteins. However, the approach faces significant challenges due to the proteome complexity in biological samples. The presence of high-abundance proteins, like albumin, can obscure low-abundance proteoforms, complicating separation techniques like liquid chromatography. Additionally, the large size of intact proteins demands advanced ion fragmentation methods and sophisticated bioinformatics for accurate data interpretation.
Workflow of Top-Down Proteomics
Conducting Top-Down Proteomics involves several critical steps, from sample preparation to data analysis. Below is a detailed guide to the entire process:
1. Sample Preparation
- Source Selection: Choose an appropriate biological sample, such as tissue, cell lysates, or body fluids. Ensure the sample is suitable for proteomic analysis.
- Protein Extraction: Isolate proteins from the sample using techniques like homogenization or centrifugation, depending on the sample type. Use buffers that maintain protein stability and prevent degradation.
- Concentration and Purification: Concentrate the protein solution using techniques such as precipitation (e.g., ammonium sulfate) or ultrafiltration to remove small molecules and contaminants.
2. Direct Protein Analysis
- Introduce Proteins to Mass Spectrometer: Load the intact proteins directly into the mass spectrometer. Ensure that the sample is in a suitable solvent that can be ionized effectively, often using a method like electrospray ionization (ESI).
- Ionization: Generate charged ions from the intact proteins for analysis. ESI is commonly used for Top-Down Proteomics because it can produce multiply charged ions from larger proteins.
3. Mass Spectrometry Identification
1) Fragmentation Techniques:
- Electron Transfer Dissociation (ETD): This method allows for the fragmentation of ions while preserving labile post-translational modifications, providing structural information about the protein.
- Ultraviolet Photodissociation (UVPD): UVPD uses high-energy ultraviolet light to induce fragmentation, offering another way to analyze protein structure.
2) Acquisition of Mass Spectra: Collect the mass-to-charge (m/z) ratios and intensity data for the intact proteins and their fragments. The mass spectrometer will produce spectra that represent the protein and its fragments.
4. Data Analysis
- Spectrum Interpretation: Use specialized software to interpret the mass spectrometry data. Analyze the mass spectra to identify protein peaks and their corresponding fragments.
- Protein Identification: Match the observed m/z values to known protein sequences using databases such as UniProt or NCBI. The identification may include assessing post-translational modifications and isoforms.
- Quantitative Analysis: If needed, use methods like label-free quantification or stable isotope labeling to compare protein abundance across different samples or conditions.
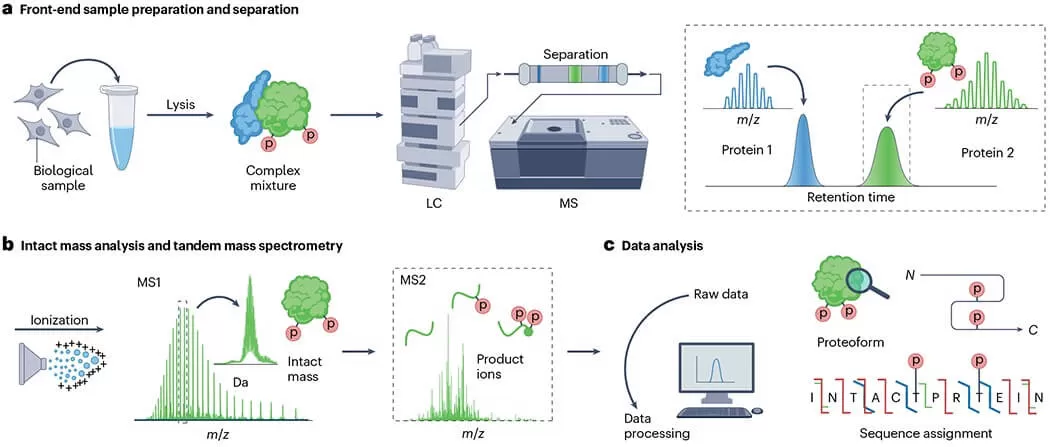
Characteristics of Top-Down Proteomics
- Preservation of Protein Integrity: Top-Down Proteomics allows for the simultaneous analysis of a protein's primary structure and post-translational modifications (PTMs). This capability provides a comprehensive view of the protein’s functional state.
- Isomer Resolution: This approach can directly differentiate between various proteoforms or isomers of a protein. By analyzing intact proteins, researchers can identify subtle differences that may significantly impact function.
- No Need for Database Searching: Proteins can be identified directly from mass spectrometry data, reducing the complexity associated with database searches. This advantage streamlines the identification process and minimizes potential errors that can arise during matching.
Limitations of Top-Down Proteomics:
- High Technical Requirements: The method demands high-resolution mass spectrometers and sophisticated data processing techniques. These requirements can pose challenges in terms of cost and accessibility, especially for laboratories with limited resources.
- Lower Analytical Throughput: Compared to Bottom-Up Proteomics, Top-Down methods generally have a lower throughput. This results in slower analysis times, making it more suitable for in-depth studies of a limited number of proteins rather than large-scale proteomic profiling.
Bottom-Up Proteomics: Fragmenting Proteins for Analysis
Bottom-up proteomics is the foundation of proteomics, of which shotgun proteomics is the most well-known. Bottom-up proteomics uses proteases such as trypsin to enzymatically digest proteins into smaller peptide fragments. These peptides are then analyzed by liquid chromatography-mass spectrometry (LC-MS), enabling high-throughput proteomic analysis of complex protein mixtures. The advantage of this approach is its scalability, making it ideal for large-scale proteome analysis in biomarker discovery and disease research. However, peptide-based analysis can lose post-translational modification (PTM) information, making the identification of protein variants more complicated.
Workflow of Bottom-Up Proteomics
Conducting Bottom-Up Proteomics involves a series of well-defined steps that encompass protein extraction, enzymatic digestion, peptide separation, and mass spectrometry analysis. Here is a comprehensive guide to the process:
1. Sample Preparation
- Sample Collection: Choose an appropriate biological sample, such as cell lysates, tissue homogenates, or body fluids. Ensure that the sample is suitable for proteomic analysis.
- Protein Extraction: Isolate proteins from the sample using methods like homogenization or centrifugation. Use a lysis buffer containing detergents and protease inhibitors to solubilize and stabilize the proteins.
- Protein Quantification: Determine the concentration of extracted proteins using methods such as the Bradford assay, BCA assay, or UV absorbance at 280 nm.
2. Enzymatic Digestion
- Select a Protease: Common proteases for digestion include trypsin, chymotrypsin, and LysC. Trypsin is the most widely used due to its specificity and efficiency.
- Digestion Process: Dilute the protein sample to reduce the concentration and facilitate digestion. Add the chosen protease in a specific enzyme-to-substrate ratio (typically 1:50 to 1:100) and incubate at an appropriate temperature (usually 37°C) for several hours or overnight. If necessary, consider performing a two-step digestion using different proteases for increased coverage of protein sequences.
3. Peptide Purification
- Stop Digestion: Once digestion is complete, stop the enzymatic reaction by adjusting the pH with formic acid or acetic acid.
- Peptide Cleanup: Remove salts, detergents, and other impurities using methods like:
- Solid-Phase Extraction (SPE): This method uses specialized columns to isolate and concentrate peptides.
- Ultrafiltration: Use membrane filters to concentrate and desalt the peptide solution.
4. Peptide Separation
Liquid Chromatography: Employ techniques such as reversed-phase liquid chromatography (RPLC) to separate peptides based on their hydrophobicity. This step reduces sample complexity and improves the quality of mass spectrometry data.
Use a gradient elution method to gradually increase the organic solvent concentration, facilitating the elution of peptides at different retention times.
5. Mass Spectrometry Analysis
- Ionization: Ionize the separated peptides using methods such as electrospray ionization (ESI) or matrix-assisted laser desorption/ionization (MALDI) to generate charged ions.
- Mass Spectrometry (MS): Introduce the ionized peptides into the mass spectrometer to measure their mass-to-charge (m/z) ratios. Collect the mass spectra for analysis.
- Tandem Mass Spectrometry (MS/MS): Select specific peptides for fragmentation to generate product ions. This provides additional information about the amino acid sequences and any post-translational modifications present.
6. Data Analysis
- Spectrum Interpretation: Use specialized software to analyze the mass spectrometry data. This includes identifying peptide sequences based on their m/z values and fragmentation patterns.
- Database Search: Match the identified peptide sequences to protein databases such as UniProt or NCBI to determine the corresponding proteins. Employ search algorithms that consider modifications and ensure accurate identification.
- Quantitative Analysis: Use label-free quantification methods or isotopic labeling techniques (such as TMT or iTRAQ) to quantify protein abundance across different samples or conditions.
Advantages of Bottom-Up Proteomics
- High Throughput and Scalability: Bottom-Up Proteomics is highly effective for analyzing large numbers of proteins in complex samples, making it ideal for high-throughput studies in various research fields.
- Established Protocols: There are well-established protocols and methodologies for Bottom-Up Proteomics, which facilitate the reproducibility and reliability of results.
- Enhanced Sensitivity: The method typically achieves higher sensitivity and precision, allowing for the detection of low-abundance proteins and a wide dynamic range of protein concentrations.
Limitations of Bottom-Up Proteomics
- Challenges in Analyzing Protein Modifications: Since proteins are digested into smaller peptide fragments, information about certain post-translational modifications (PTMs) may be lost during the digestion process. This limitation can hinder the ability to fully characterize the functional state of proteins.
- Limited Resolution of Protein Isoforms: Different isoforms of a protein may produce identical peptide fragments during digestion, making it challenging to distinguish between them. This limitation can lead to ambiguities in identifying specific proteoforms, which are crucial for understanding the diversity and function of proteins in biological systems.
Top-Down vs. Bottom-Up Proteomics: Comparative Summary
- Analysis Workflow: Bottom-Up Proteomics begins with the digestion of proteins into peptide fragments, while Top-Down Proteomics involves the direct analysis of intact proteins.
- Data Type: Bottom-Up Proteomics provides information at the peptide level, whereas Top-Down Proteomics yields comprehensive data about the entire protein.
- Application Areas: Bottom-Up methods are well-suited for large-scale proteomic screening, while Top-Down methods are ideal for structural and modification analyses of proteins.
- Technical Difficulty: Top-Down Proteomics requires more advanced technical capabilities, resulting in more complex data analysis.
Both methodologies have their unique advantages, and the choice between them depends on the specific research objectives and requirements. As technology advances, these two approaches may further integrate, offering a more comprehensive analytical framework for proteomics research.
Conclusion
Some researchers have proposed integrating Top-Down and Bottom-Up methods for proteomic analysis. This hybrid strategy allows for complementary strengths across different levels of analysis, such as utilizing Top-Down approaches to detect degradation products and sequence variations, while employing Bottom-Up techniques for large-scale peptide identification and quantification.
In summary, both Top-Down and Bottom-Up Proteomics possess distinct advantages and limitations. The selection of a particular strategy depends on the specific research needs and goals. For instance, when high sensitivity and accurate localization of post-translational modifications are required, Top-Down strategies may be more advantageous. Conversely, Bottom-Up approaches are often more suitable for efficiently obtaining proteomic information and handling complex samples.
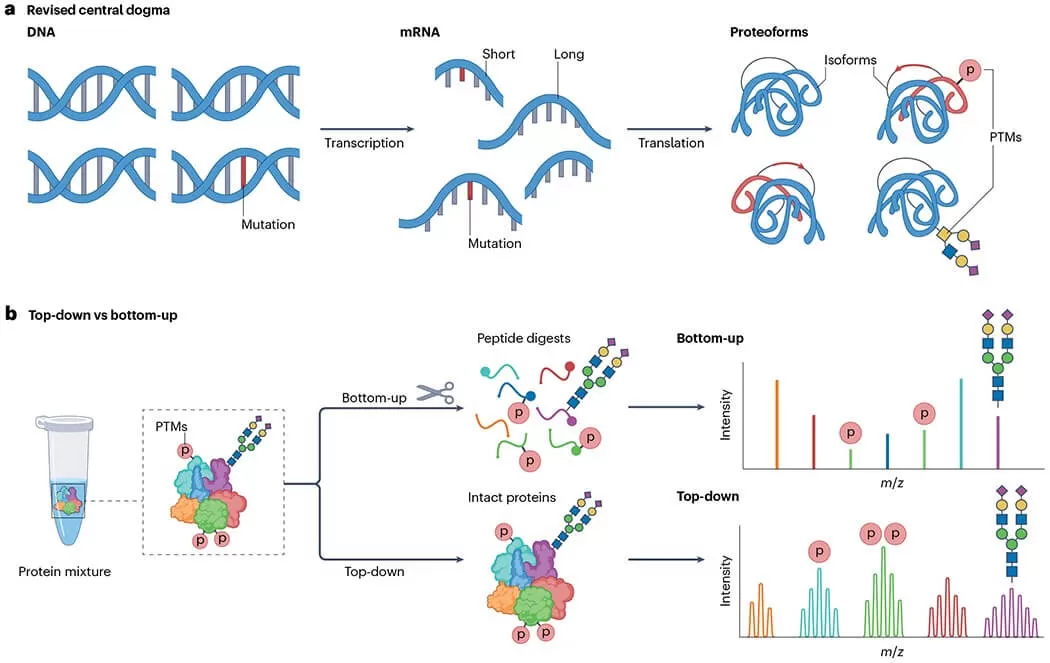
FAQ
Q1: What is top-down vs bottom-up data analysis?
In proteomics, top-down and bottom-up data analysis differ in approach and output. Bottom-Up Proteomics digests proteins into peptide fragments for liquid chromatography-mass spectrometry (LC-MS) analysis, enabling high-throughput proteome profiling but losing post-translational modification (PTM) context. Top-Down Proteomics analyzes intact proteins, preserving proteoforms and PTM details with high-resolution MS, but it’s less scalable. Bottom-Up suits broad screening; Top-Down excels in precise proteoform characterization.
Q2: What are the benefits of proteomics?
Proteomics reveals dynamic protein functions, offering key benefits in research and industry. It enables protein biomarker discovery for early disease detection, supports precision medicine via pharmacoproteomics, and identifies drug targets by mapping disease pathways. By integrating with genomics and metabolomics, proteomics provides holistic systems biology insights.
Q3: What is top down proteomics?
Top-Down Proteomics analyzes intact proteins using high-resolution mass spectrometry without digestion, preserving proteoforms and post-translational modifications (PTMs). It uses techniques like electron-transfer dissociation (ETD) to fragment proteins, enabling precise PTM localization and proteoform identification. Ideal for small proteins and disease research, it offers fewer false positives but faces challenges with large proteins and lower throughput.
Reference
Gregorich, Z. R., Chang, Y. H., & Ge, Y. (2014). Proteomics in heart failure: top-down or bottom-up? Pflugers Archiv: European journal of physiology, 466(6), 1199–1209. https://doi.org/10.1007/s00424-014-1471-9
Roberts, D. S., Loo, J. A., Tsybin, Y. O., Liu, X., Wu, S., Chamot-Rooke, J., Agar, J. N., Paša-Tolić, L., Smith, L. M., & Ge, Y. (2024). Top-down proteomics. Nature reviews. Methods primers, 4(1), 38. https://doi.org/10.1038/s43586-024-00318-2
Next-Generation Omics Solutions:
Proteomics & Metabolomics
Ready to get started? Submit your inquiry or contact us at support-global@metwarebio.com.
