
TMT vs. DIA: Which Method Reigns Supreme in Quantitative Proteomics?
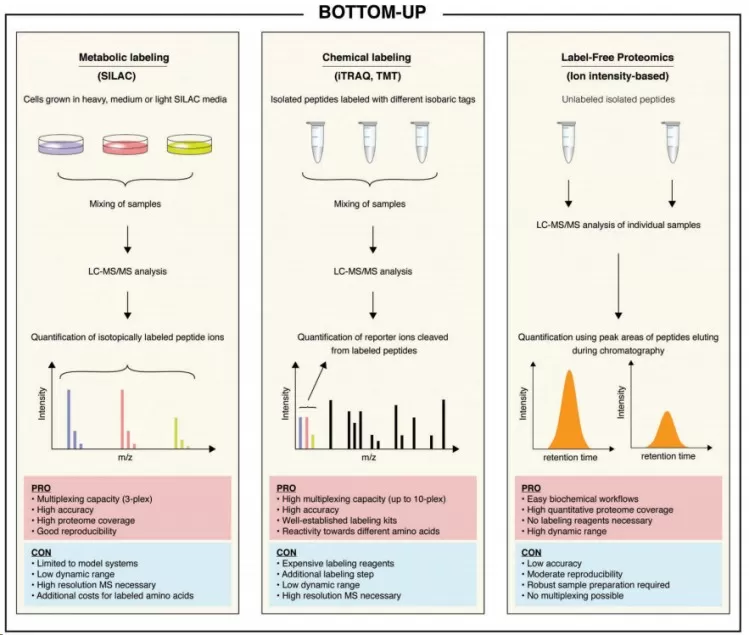
Currently, bottom-up mass spectrometry has become the most important and popular tool for proteomic research, widely applied in the identification, quantification, and characterization of proteins and protein modifications. In bottom-up proteomics, protein samples are digested into peptide fragments, which are then analyzed using liquid chromatography-mass spectrometry (LC-MS). The resulting data undergoes analysis to provide qualitative and quantitative information about the proteins. With this method, quantitative proteomics can be categorized into two main approaches based on sample preparation: labeled quantitative proteomics and label-free quantitative proteomics.
Labeled quantitative proteomics typically involves using chemical tags or labels that are attached to peptides, enabling the relative quantification of proteins across different samples. This approach allows for more precise comparisons between conditions due to its ability to standardize measurements across analytical runs. Among the various labeling techniques, TMT (Tandem Mass Tag) labeling has emerged as one of the most representative and widely adopted methods in quantitative proteomics.
In contrast, label-free quantitative proteomics operates without the need for labeling techniques. Instead, it quantifies proteins by analyzing the intensity of detected peptides, enabling a straightforward assessment of complex biological samples. This approach is especially beneficial in high-throughput studies, as it streamlines sample preparation and reduces costs. A leading method in this category is DIA (Data-Independent Acquisition) quantitative proteomics.
Given the unique features and technical strengths of both approaches, many researchers face challenges when deciding which method to employ for their studies. In this blog, we will comprehensively explore the differences between labeled quantitative proteomics and label-free quantitative proteomics, using TMT and DIA (Data-Independent Acquisition) as key examples. Our aim is to provide researchers with the insights needed to select the most suitable techniques for their specific research objectives.
Differences in Technical Principles
Quantification Principle of TMT Technology
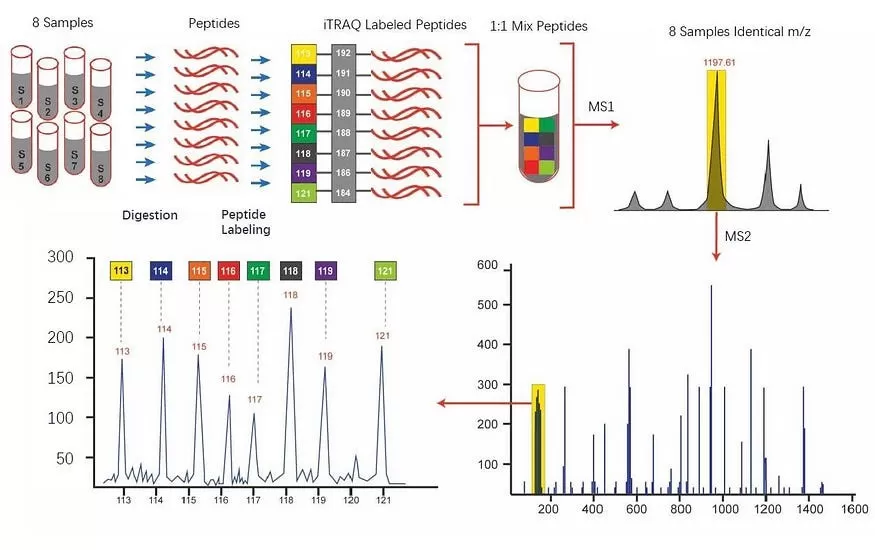
TMT (Tandem Mass Tag) quantitative proteomics involves labeling peptide segments derived from protein digests in different samples using multiple isotopic tag reagents. These labeled peptides are then mixed together for mass spectrometry (MS) analysis. Since peptides with the same sequence display identical mass-to-charge ratios (m/z) due to the uniformity of the tags, they appear as a single peak in the MS1 spectrum. When this peak is selected for fragmentation, each isotopic tag generates a unique reporter ion profile in the MS2 spectrum. By comparing the intensities of the different reporter ions, relative quantification of the same protein across samples can be achieved.
Quantification Principle of DIA Technology
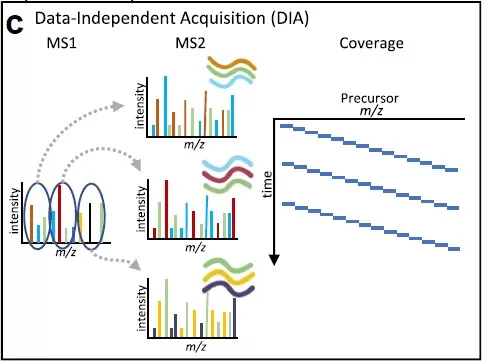
In contrast, DIA (Data-Independent Acquisition) quantitative proteomics eliminates the need for costly isotopic labeling. Instead, after extracting and digesting proteins from samples, peptides are analyzed directly via LC-MS using a data-independent acquisition mode. During the mass spectrometry data collection, an initial full MS1 scan captures information on parent ions within a defined time window, recording their m/z ratios and intensities. Subsequently, this scanning window is divided into several narrower windows based on the m/z ratios of the parent ions. During the MS2 scan, specific peptide segments are selected using these predefined wide mass windows and fragmented in the collision cell. This process continues iteratively until MS2 spectra for all parent ions are produced, enabling quantitative analysis based on the intensity of the peptide fragments.
Differences in Experimental Procedures
TMT Quantitative Proteomics Workflow
The workflow for TMT quantitative proteomics begins with protein extraction and digestion, followed by the labeling of peptides from different samples using isotopic tags. These labeled peptides are then combined. Given the complexity of the mixed peptide information, chromatography is employed to fractionate the mixture into different fractions for individual mass spectrometry analysis. The greater the number of fractions, the deeper the protein coverage achieved.

DIA Quantitative Proteomics Workflow
The workflow for DIA quantitative proteomics involves total protein extraction from the sample, followed by enzymatic digestion with trypsin to produce peptides. Each sample’s peptides are then analyzed directly using mass spectrometry in the DIA mode for data collection. The qualitative and quantitative analysis is performed based on the MS2 spectra and the corresponding intensity responses of the peptide fragments.
Different Applicable Scenarios
The experimental workflow for TMT quantitative proteomics involves labeling the samples, mixing them, and then fractionating them for mass spectrometry. This method allows for the simultaneous detection of the same peptide segment across multiple samples, leading to high protein detection rates and deep coverage, with good quantification stability. Typically, samples from the same tissue or cell type exhibit similar protein profiles, making them well-suited for TMT analysis.
However, TMT kits are currently limited to a maximum of 18 tags. When dealing with more than 18 samples, multiple kits must be used, which introduces batch effects. Additionally, the labeling efficiency may vary across different tissues. Therefore, for experiments involving large sample sizes or diverse tissue types, DIA quantitative proteomics is recommended, as it ensures both deep protein coverage and stable quantification.
Comprehensive Comparison of TMT and DIA
|
Technique |
TMT Quantitative Proteomics |
|
|
Scanning Mode |
DDA |
DIA |
|
Identification Level |
MS2 |
MS2 |
|
Quantification Level |
MS2 |
MS2 |
|
Advantage |
Better reproducibility |
Wider application range and lower cost |
|
Limitation |
Limited sample types, small sample sizes, high costs |
complex data processing |
Next-Generation Omics Solutions:
Proteomics & Metabolomics
Ready to get started? Submit your inquiry or contact us at support-global@metwarebio.com.
