
From Data to Discovery: Protein Identification and Quantification in MS-based Proteomics
In proteomics, the journey from raw data to meaningful biological insights relies heavily on accurate protein identification and quantification. Mass spectrometry (MS)-based techniques are at the heart of this process, enabling researchers to delve deep into the complexities of protein expression, modifications, and interactions. However, transforming the vast amount of data generated by MS into reliable protein profiles requires a structured approach, combining sophisticated algorithms, database searching, and quantification methods.
In this blog, we will explore how mass spectrometry data is processed to identify proteins, the principles behind quantifying protein abundance, and the key strategies used in modern proteomics for extracting accurate, reproducible information from complex biological samples. Whether you're new to proteomics or looking to refine your understanding, this guide will provide insights into the essential workflows that turn raw data into actionable discoveries.
Peptide Mass Fingerprinting (PMF) Protein Identification Strategy
Peptide mass fingerprinting (PMF) is a technique used to identify proteins based on the unique mass data of their peptide fragments. After digesting proteins with specific endoproteases, peptides of distinct lengths and masses are produced. These peptides form a unique signature, known as the peptide mass fingerprint. The steps involved in PMF are shown in Figure 1. However, PMF results can sometimes lack specificity and clarity due to various influencing factors. The advent of tandem mass spectrometry (MS/MS) has significantly advanced proteomics by providing more detailed peptide information through peptide fragmentation.

In an MS/MS experiment, the mass spectrometer first captures all peptide ion information at a given time point (MS1 full scan), measuring all intact peptide ions present. Selected peptides are then isolated, fragmented, and the resulting fragment ions are measured to generate an MS/MS spectrum. This process is repeated throughout the LC gradient, automatically capturing as many different peptide ions as possible. Figure 2 illustrates the relationship between precursor ions in the MS scan and their corresponding MS/MS spectra.

While numerous search engines are available to analyze MS/MS spectra (Table 1), most follow a similar identification strategy. First, theoretical digestion is performed on protein sequences, generating theoretical peptide fragments. Specific modifications (such as adding specific mass groups to certain amino acids) are simulated, and theoretical fragmentation spectra are produced for each peptide. These theoretical spectra are then compared and scored against experimental MS/MS spectra. Each experimental spectrum is matched with the highest-scoring peptide sequence, and the matched peptides are filtered to identify the corresponding proteins (Figure 3).
Table 1. Mainstream Proteomics Search Engines
|
Index |
Software |
|
1 |
|
|
2 |
|
|
3 |
|
|
4 |
|
|
5 |
|
|
6 |

Protein Identification Using DIA Data
In Data-Dependent Acquisition (DDA), only a subset of precursor ions is selected for fragmentation based on signal strength, which leads to a loss of many precursor ions. In contrast, Data-Independent Acquisition (DIA) uses predefined isolation windows to continuously fragment all precursor ions within a mass range of interest, without prioritizing the most abundant ions. This approach captures nearly all ion information, resulting in high data reproducibility and stability.However, DIA’s wide isolation windows lead to complex spectra with overlapping fragment ions, making deconvolution of these spectra crucial and challenging in DIA data analysis.
DIA analysis software generally falls into two categories: Library-Based (dependent on DDA-generated libraries) and Library-Free (without a pre-existing library). A spectral library typically consists of precursor ion m/z, fragment ion m/z, relative intensities, and retention times. Advanced libraries may also include ion mobility data, peptide charge states, specific fragments, and optimal elution times. The library is used to extract target peptides from the DIA data, and the target-decoy FDR method evaluates the identification’s accuracy by scoring factors like mass error and RT deviation.
In library-based DIA, only peptides present in the library can be identified. Missing or low-abundance peptides can decrease sensitivity and specificity, while increasing computational demands. The library’s size, composition, and quality of fragment ion data directly influence the accuracy of protein quantification and biological inference.
Library-Free methods, also known as spectrum-centric approaches, perform untargeted analysis of DIA data without a pre-existing library. Tools like DIA-Umpire detect precursor and fragment ion features to create pseudo MS/MS spectra, which can be analyzed using traditional protein identification tools. Alternatively, peptide-centric methods like PECAN use peptide and protein background data (typically from a species database) to assign scores based on peptide identification evidence. Percolator then uses these scores to estimate false discovery rates and report reliable peptide and protein identifications.
Protein Quantification Strategies
Mass spectrometry (MS) is a versatile tool for measuring the mass of ionized molecules. In proteomics, MS is utilized across various fields, each requiring tailored analytical approaches, instrumentation, and data processing steps. To accommodate different experimental needs, a range of algorithms and software for quantitative proteomics has emerged.
Protein quantification methods are primarily divided into two categories based on isotope labeling:
- Label-free methods like SWATH and DIA.
- Labeled methods such as TMT, iTRAQ, SILAC, and ICAT.
Quantification is further classified by the source of information:
- MS1-based quantification (precursor ions), used in label-free, SILAC, and ICAT methods.
- MS2-based quantification (fragment ions), seen in SWATH and DIA.
- Reporter ion-based quantification, as in iTRAQ and TMT, relies on reporter ions in MS2 spectra.
Additionally, strategies are categorized by acquisition mode:
- Data-Dependent Acquisition (DDA), used in most traditional methods.
- Data-Independent Acquisition (DIA), a label-free, non-selective approach that captures all precursor ions for comprehensive quantification.
Label-Free Quantification (LFQ) Methods
Label-free quantification methods are divided into two categories based on different quantification principles (Figure 3):
1) Extracted Ion Chromatogram (XIC) Method: This approach quantifies peptides by extracting the mass-to-charge ratios (m/z) of precursor ions across retention time (RT) to generate an XIC. The area under the XIC or the summed signal intensity is used for peptide quantification. There are two ways to identify the peptide sequences corresponding to the XIC:
(i) Matching with the Accurate Mass and Time (AMT) database.
(ii) Identifying peptides using results from a protein sequence database search.
2) Spectral Counting (SC): This method quantifies proteins based on the principle that higher protein abundance increases the likelihood of detecting more peptide spectra. The number of peptide spectrum matches (PSMs) associated with a protein is used to infer its relative abundance.

Isotopic Labeling Quantification in Proteomics
Stable isotopic labeling is a method where samples are labeled with stable isotopes, allowing for identification and quantification through mass spectrometry. The core principle is that peptides in different labeling states form isotope peaks with fixed mass differences, which are used to calculate peptide abundance and ratios. Based on the labeling target, this method is divided into two types:
1) Precursor Ion Labeling: Isotopic labels are introduced at the precursor ion level, creating mass differences in MS1 spectra.
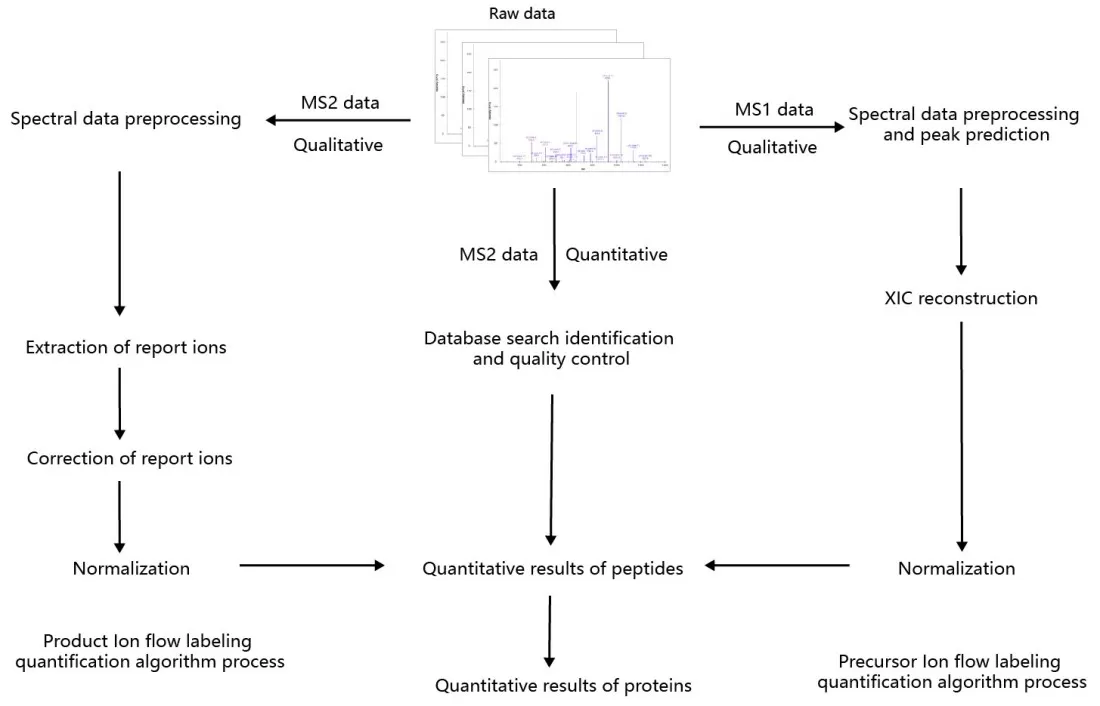
2) Fragment Ion Labeling: Isotopic labels are introduced at the fragment ion level, creating mass differences in MS2 spectra (Figure 4).
Reference:
1. Mann, M. and M. Wilm, Error-tolerant identification of peptides in sequence databases by peptide sequence tags. Anal Chem, 1994. 66(24): p. 4390-9.
2. Thiede, B., et al., Peptide mass fingerprinting. Methods, 2005. 35(3): p. 237-47.
3. Eng, J.K., et al., A face in the crowd: recognizing peptides through database search. Mol Cell Proteomics, 2011. 10(11): p. R111 009522.
4. Meier, F., et al., diaPASEF: parallel accumulation-serial fragmentation combined with data-independent acquisition. Nat Methods, 2020. 17(12): p. 1229-1236.
5. Elias, J.E. and S.P. Gygi, Target-decoy search strategy for increased confidence in large-scale protein identifications by mass spectrometry. Nat Methods, 2007. 4(3): p. 207-14.
6. Tsou, C.C., et al., DIA-Umpire: comprehensive computational framework for data-independent acquisition proteomics. Nat Methods, 2015. 12(3): p. 258-64, 7 p following 264.
7. Ting, Y.S., et al., Peptide-Centric Proteome Analysis: An Alternative Strategy for the Analysis of Tandem Mass Spectrometry Data. Mol Cell Proteomics, 2015. 14(9): p. 2301-7.
8. Ting, Y.S., et al., PECAN: library-free peptide detection for data-independent acquisition tandem mass spectrometry data. Nat Methods, 2017. 14(9): p. 903-908.
9. Kall, L., et al., Semi-supervised learning for peptide identification from shotgun proteomics datasets. Nat Methods, 2007. 4(11): p. 923-5.
10. Strittmatter, E.F., et al., Proteome analyses using accurate mass and elution time peptide tags with capillary LC time-of-flight mass spectrometry. J Am Soc Mass Spectrom, 2003. 14(9): p. 980-91.
11. Lundgren, D.H., et al., Role of spectral counting in quantitative proteomics. Expert Rev Proteomics, 2010. 7(1): p. 39-53.
Next-Generation Omics Solutions:
Proteomics & Metabolomics
Ready to get started? Submit your inquiry or contact us at support-global@metwarebio.com.
