
Mastering Western Blot: A Step-by-Step Guide from Sample to Signal
Western blotting (WB) is a molecular biology technique used to detect the expression levels of specific proteins in a sample. It is a widely employed experimental method in molecular biology, biochemistry, and immunogenetics. The fundamental principle of Western blotting relies on the separation of proteins by SDS-PAGE (sodium dodecyl sulfate polyacrylamide gel electrophoresis) and the specific recognition of proteins by antibodies. Through colorimetric or chemiluminescent detection techniques, this method enables the identification of the target protein.
The experimental workflow consists of several key steps: total protein extraction, SDS-PAGE electrophoresis, membrane transfer, signal detection, and data analysis. Each step is critical to ensuring the specificity and sensitivity of the protein detection process.
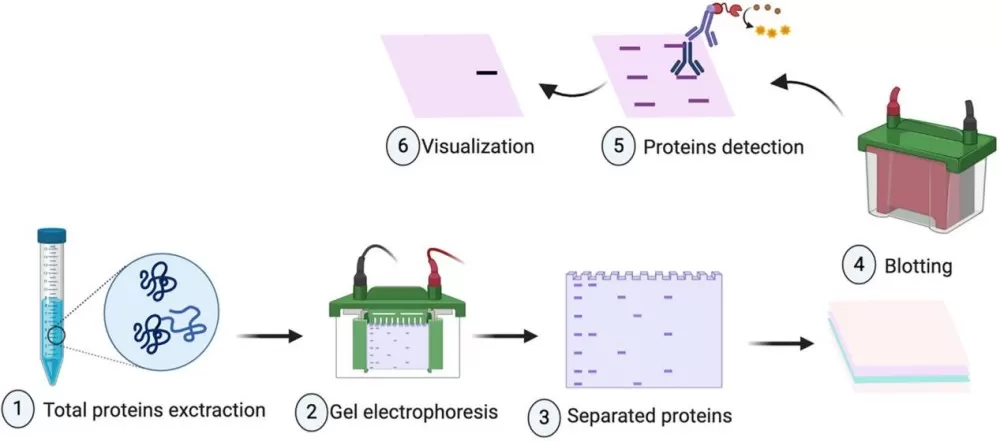
1. Total Protein Extraction:
1) Sample Preparation: Tissue Samples-Cut the tissue into small pieces, place them in a pre-chilled mortar, rapidly freeze using liquid nitrogen, and then grind into a powder with a pestle. Cell Samples-Culture cells to the desired density, remove the culture medium, and wash the cells 1-2 times with pre-chilled PBS.
2) Lysis: Transfer the ground tissue powder or cells into a pre-chilled centrifuge tube. Add a pre-chilled lysis buffer (containing protease and phosphatase inhibitors such as SDS, Tris-HCl, PMSF, and EDTA) and mix thoroughly using a pipette.
3) Incubation: Place the centrifuge tube on ice and gently vortex or pipette up and down every 5-10 minutes to aid in lysis. For cell samples, lysis typically lasts 10-30 minutes, while for tissue samples, it may take longer, around 30 minutes to 1 hour.
4) Centrifugation: Centrifuge the lysed samples at 4°C, usually at 12,000-15,000 rpm for 10-15 minutes. After centrifugation, transfer the supernatant (containing total protein) to a new pre-chilled centrifuge tube.
5) Protein Quantification: Quantify the extracted proteins using methods such as BCA or Bradford to ensure equal loading of protein samples in subsequent experiments.
6) Protein Denaturation: Mix the protein samples with loading buffer containing SDS and a reducing agent (such as DTT or β-mercaptoethanol), then heat in boiling water (99°C for 10 minutes) to denature the proteins in preparation for electrophoresis.
Note:
a) Ensure complete lysis of proteins to avoid freeze-thaw cycles.
b) Select an appropriate lysis buffer, such as RIPA lysis buffer, and include protease inhibitors to prevent protein degradation.
c) Maintain low temperatures during sample preparation to minimize protein degradation.
2. SDS-PAGE Electrophoresis
1) Sample Loading: Load the denatured protein samples into the polyacrylamide gel, and place the gel into an electrophoresis apparatus filled with running buffer.
2) Protein Separation: Separate proteins based on their molecular weight under the influence of an electric field. Stop the electrophoresis when the target bands reach the midpoint of the separating gel.
Notes:
a) Choose an appropriate gel concentration for separating proteins of different sizes; higher concentrations are suitable for smaller proteins, while lower concentrations are better for larger proteins.
b) Ensure thorough mixing of samples with the loading buffer and perform electrophoresis at appropriate voltage levels to avoid excessive heat, which can denature proteins; typically, a low voltage of 60V is recommended.
c) 1× Running Buffer Composition: 3 g Tris base, 14.5 g Glycine, 5 ml 20% SDS, adjusted to 1 L with ddH₂O.
d) Separation Gel Preparation (12%): 1.6 ml ddH₂O, 2 ml 30% acrylamide, 1.3 ml 1.5 M Tris (pH 8.8), 0.05 ml 10% SDS, 0.002 ml TEMED, 0.05 ml 10% APS. Concentration Gel Preparation (12%): 1.4 ml ddH₂O, 0.33 ml 30% acrylamide, 0.25 ml 1.5 M Tris (pH 6.8), 0.02 ml 10% SDS, 0.002 ml TEMED, 0.02 ml 10% APS.
3. Membrane Transfer
1) Transfer: Transfer the separated proteins from the gel to a solid support membrane (typically nitrocellulose or PVDF). This can be achieved through wet transfer (for proteins >100 kDa) or semi-dry transfer (for smaller proteins). For semi-dry transfer, stack thick filter paper, thin filter paper, membrane, protein gel, thin filter paper, and thick filter paper in the transfer apparatus. Moisten each layer with TGM solution, and use a glass rod to eliminate air bubbles (ensure filter paper and membrane are pre-soaked). The transfer time depends on the protein's molecular weight: for proteins <50 kDa, use 24V for 30 minutes; for proteins between 50 kDa and 100 kDa, use 24V for 30-60 minutes.
2) Blocking: Treat the membrane with a blocking solution containing milk or non-fat dry milk to prevent non-specific binding.
3) Primary Antibody Incubation: Wash the membrane with TBST solution, then incubate with a specific primary antibody targeting the protein of interest, allowing the antibody to bind to the target protein.
4) Secondary Antibody Incubation: After washing away unbound primary antibody with TBST, incubate the membrane with a secondary antibody labeled with an enzyme or fluorophore, which targets the primary antibody.
Notes:
a) Choose the appropriate transfer method (wet, semi-dry, or dry). Transfer efficiency is influenced by factors such as protein size, transfer time, and voltage; larger proteins may require overnight transfer, while smaller proteins may need shorter transfer times.
b) 1× TGM Composition: 1.514 g Tris base, 7.206 g Glycine (Bio-Rad), 100 ml Methanol, adjusted to 500 ml with ddH₂O.
c) Use an appropriate blocking solution (e.g., 5% non-fat dry milk) to block unbound sites on the membrane, typically for at least 1 hour or overnight.
d) Select suitable primary and secondary antibodies and follow recommended dilution ratios. Primary antibody incubation can be performed at room temperature for 2 hours or overnight at 4°C; secondary antibody incubation is generally at room temperature for 1 hour.
e) After incubating with the primary and secondary antibodies, wash thoroughly with TBST. Washing is crucial for accurate experimental results; use freshly prepared 1× TBST (composition: 7.3 g NaCl, 3.028 g Tris base, 1 ml Tween 20, pH 8.0, adjusted to 1 L with ddH₂O) for 5 minutes each wash. If background signals are high, consider increasing wash frequency and duration.
4. Signal Detection and Data Analysis
1) Signal Detection: Wash the membrane again with TBST, then use chemiluminescent (ECL) or fluorescent methods to detect the signal from the secondary antibody, capturing the band images with imaging equipment.
2) Data Analysis: Utilize image analysis software (such as ImageJ or Quantity One) to scan and analyze the bands, allowing for quantitative comparison of the target protein expression levels across different samples.
Notes: If issues arise, such as the absence of bands, weak bands, high background, or incorrect band sizes, investigate the antibodies, lysis buffer, transfer efficiency, blocking effectiveness, and antibody incubation conditions.
Reference:
Meftahi GH, Bahari Z, Zarei Mahmoudabadi A, Iman M, Jangravi Z. Applications of western blot technique: From bench to bedside. Biochem Mol Biol Educ. 2021 Jul;49(4):509-517. doi: 10.1002/bmb.21516. Epub 2021 Apr 13. PMID: 33847452.
Next-Generation Omics Solutions:
Proteomics & Metabolomics
Ready to get started? Submit your inquiry or contact us at support-global@metwarebio.com.
